

Cool, very useful if you want to batch-query many accession numbers. You might want to add an option like --linux and --osx to output the correct path to the default aspera openssh file as default paths are a bit different in both operating systems. If you want to search NCBI also check out sra-explorer : find SRA and FastQ download URLs in a couple of clicks from Phil Ewels which has an option to print ENA links directly.
Tutorial: [Deprecated] Fast download of FASTQ files from the European Nucleotide Archive (ENA)
Info: 28.6.22
This tutorial is all-deprecated. Use sra-explorer.info to get download links from ENA and Setting up Aspera Connect (ascp) on Linux and macOS to learn how to setup Aspera.
Info: 16.05.2021
The layout of ENA changed so the steps in this tutorial towards finding and selecting datasets are deprecated, and I have not yet found the time to update it. Actually, as there is sra-explorer these days I recommend to just use this to query ENA (or ncbi), see below.
Edit 06.05.20: I actually recommend to use sra-explorer these days to get download links. This is the most convenient and fastest way. It implements the creation of download links from both NCBI and ENA including links for Aspera-driven downloads based on this tutorial. Step 1 of this tutorial can help setting up Aspera. The other steps are redundant if using sra-explorer, but this tutorial has been written before that tool was published. For more details see also sra-explorer : find SRA and FastQ download URLs in a couple of clicks and visit sra-explorer.info. It is self-explanatory.
TUTORIAL
Published sequencing data are commonly stored at NCBI and questions on how to efficiently download these data are posted frequently at Biostars. While NCBI relies on its SRA format to share these data it is possible to directly download them in fastq format from the European Nucleotide Archive (ENA1) which mirrors NCBI.
In this tutorial, we will examplarily download an entire dataset of ChIP-seq and ATAC-seq data, requiring minimal preprocessing work. We will use the Aspera client for download rates of several tens of Mb/s up to few hundred Mb/s (depending on the connection, I/O capacity and distance to the download location). This example code should work on Linux and Mac.
Step-1: Get the Aspera client
Go to https://www.ibm.com/aspera/connect/ and get the most recent installer for your system.
For Linux, it is a tarball (use tar zxvf to unpack) with an installer batch script and for Mac, a standard disk image.
After installation, there now will be these executables/files in their default locations:
Linux:
$HOME/.aspera/connect/bin/ascp --- the executable
$HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh --- openssh file that we'll need later
Mac:
$HOME/Applications/Aspera\ Connect.app/Contents/Resources/ascp --- the executable
$HOME/Applications/Aspera\ Connect.app/Contents/Resources/asperaweb_id_dsa.openssh --- openssh file that we'll need later
In any case, make sure you add the folder with the ascpexecutable to your PATH.
If PATH is a new word to you, please google it ;-)
Step-2: Choose your dataset
We have multiple options here.
Use sra-explorer: The sra-explorer is a convenient GUI to get browse NCBI for datasets. In our case we enter our accession number PRJNA288801 into the search field, then select the desired samples, add them to the cart and then copy the Aspera download links it produces. I will not add further details here as the tool is self-explainatory. It includes many (download) options, check it out. Thanks to Phil Ewels for this awesome tool!
Alternatively, query ENA / NCBI manually to find datasets: Once you know which data you want to download, check if they are backed up on the ENA, which is true for most unrestricted data. For this tutorial, we will download the entire dataset from the ChIPmentation paper of 2015. When you check the paper for the NCBI accession, you'll find GSE70482. Following this link, you find the BioSample accession number PRJNA288801. So you go to the ENA, enter this PRJNA288801 in the search field and find a summary page with all available data for download. Scrolling down a bit, you see a table with accession numbers and all kinds of metadata. As typically we do not need most of these metadata, we use the field Select columns to select the essential metadata we need for the download, which are Study Accession, FASTQ files (FTP) and Experiment title. After selecting these, and unselecting everything else, you press TEXT and save the file as accessions.txt in your project folder.
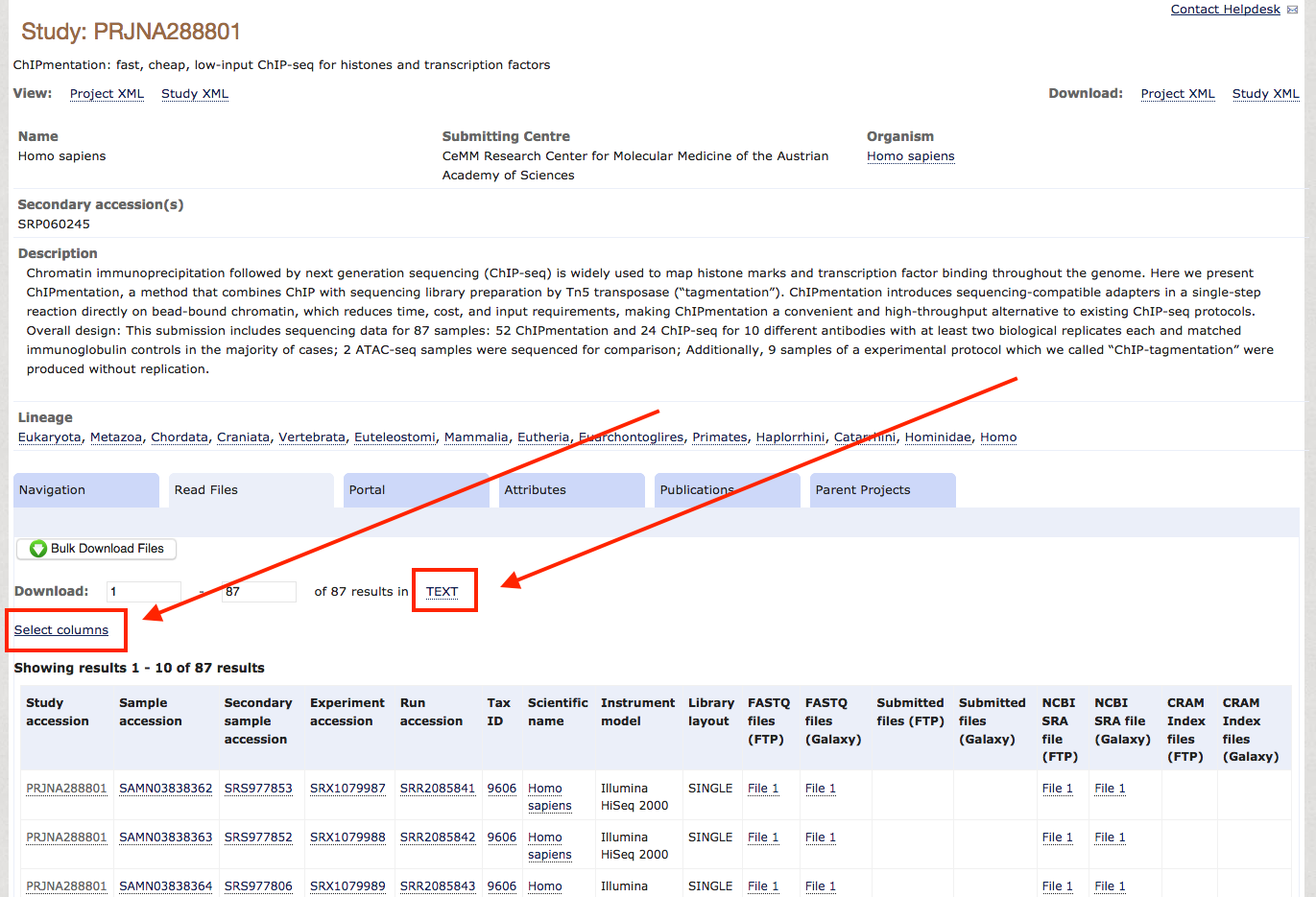
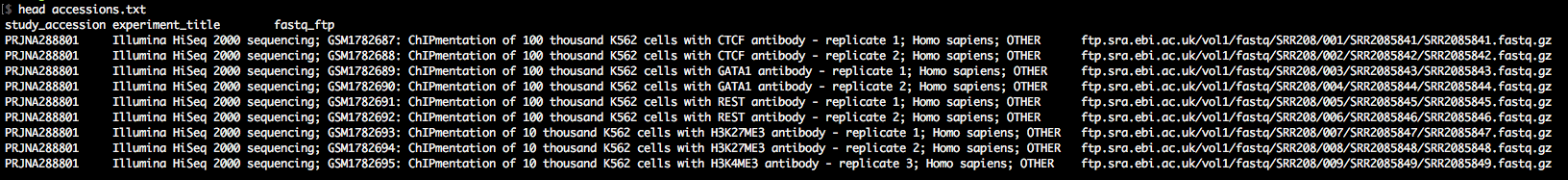
Step-3: Download the data
As you'll see in accessions.txt, the download paths direct you to the ENA ftp-server, which is rather slow. We want to download with the Aspera client (up to 200Mb/s at my workplace). Therefore, we awk around a bit to change the download paths to the era-fasp server. As you'll see in case of paired-end data, the paths to the two mate fastq files in accessions.txt
are separated by semicolon, which we take into account. The output of this snippet is download.txt.
Linux:
awk 'FS="\t", OFS="\t" { gsub("ftp.sra.ebi.ac.uk", "era-fasp@fasp.sra.ebi.ac.uk:"); print }' accessions.txt | cut -f3 | awk -F ";" 'OFS="\n" {print $1, $2}' | awk NF | awk 'NR > 1, OFS="\n" {print "ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh" " " $1 " ."}' > download.txt
Mac:
awk 'FS="\t", OFS="\t" { gsub("ftp.sra.ebi.ac.uk", "era-fasp@fasp.sra.ebi.ac.uk:"); print }' accessions.txt | cut -f3 | awk -F ";" 'OFS="\n" {print $1, $2}' | awk NF | awk 'NR > 1, OFS="\n" {print "ascp -QT -l 300m -P33001 -i $HOME/Applications/Aspera\\ Connect.app/Contents/Resources/asperaweb_id_dsa.openssh" " " $1 " ."}' > download.txt
The output is a simple list of download commands using ascp.
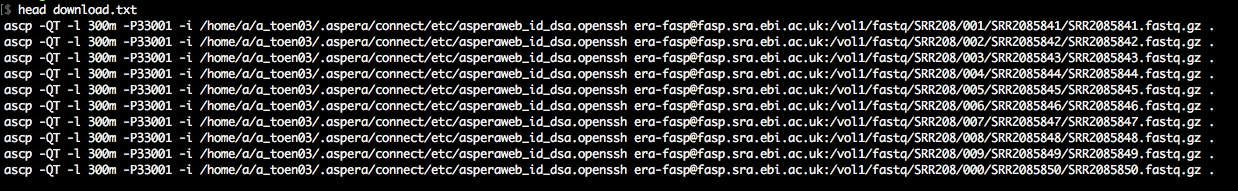
That's it. Now, we only have to run the download commands.
Edit (23.07.18): The download paths are always like era-fasp@fasp.sra.ebi.ac.uk:/vol1(...). I point that out because of a recent post (328182) where OP accidentally forgot the ":" after the .ac.uk and used fasp@ instead of era-fasp@.
Lets download:
## Either by a simple loop:
while read LIST; do
$LIST; done < download.txt
## or by using GNU parallel to have things parallelized:
cat download.txt | parallel "{}"
Once the download is complete, one can play around using the accessions.txt to rename the files with e.g. information from the Experiment title field (column 2), or other metadata you may retrieve from ENA.
Edit 28.2.19: For matters of completeness, I also add a suggestion on how to get the same data from NCBI using prefetch and parallel-fastq-dump, a wrapper for fastq-dump from Renan Valieris for parallelized fastq conversion from sra files. Say one has a file IDs.txt which contains the SRA file IDs like:
SRRXXXXXX1
SRRXXXXXX2
(...)
SRRXXXXXXn
one can use this simple function to download SRA files via prefetch, followed by fastq conversion with parallel-fastq-dump.
Note In the past prefetch could be coupled with Aspera for fast downloads, this is no longer the case, see https://github.com/ncbi/sra-tools/issues/255. Now prefetch will always download via the standard https connection.
function LoadDump {
prefetch -O ./ -X 999999999 $1
if [[ -e ${1}.sra ]]; then
parallel-fastq-dump -s ${1}.sra -t 8 -O ./ --tmpdir ./ --split-3 --gzip && rm ${1}.sra
else
echo '[ERROR]' $1 'apparently not successfully loaded' && exit 1
fi
}; export -f LoadDump
cat IDs.txt | parallel -j 2 "LoadDump {}"
This would use 8 threads for fastq conversion and run two SRA files at a time via GNU parallel, hence requiring 16 threads. As always, scale up or down based on the available resources and potential I/O bottlenecks on your system.
3 answers
Hi,
This is a very helpful post - thanks a lot for writing it. I wrote a simple Python script based on this which automates things so you only need to provide a run identifier as an argument and it works out the rest - hopefully someone will find it useful.
• 0 views
•
link
written
by
benjwoodcroft

Good idea - I added an --ssh_key option along those lines. I've not had a chance to test it on OSX though - if that is straightforward for you would you mind giving it a crack please?
• 0 views
•
link
written
by
benjwoodcroft

great tool, super useful and works like a charm, kudos!
• 0 views
•
link
written
by
andreipro87 •

The sra-explorer is currently not working for me, unfortunately.
However, when selecting the columns at the ENA website (above in Step-2: Choose your dataset), it is now possible to select fastq_aspera links directly, as shown below.
As far as I can tell, he rest of this tutorial is still up to date. Thank you for saving me from fasterq-dump that has been just giving me headache so far!!

• 0 views
•
link
written
by
Brunox13 •

Hello, first of all thank you very much for this post that I'm sure will help a lot of users ! I wondered if you could help me with an issue quite similar but for multiple genome assembly project. Here is the idea:
I have around 50 genomes assembly for wich I have their corresponding accession number in a list : Genomic_accession_and_species_names.txt
with this list I can download each SRA accession number with:
awk '{print "esearch -db assembly -query", $1, "| elink -target bioproject | elink -target sra | efetch -format docsum | xtract -pattern DocumentSummary -ACC @acc -block DocumentSummary -element \"&ACC\" | cut -f5 >>", "Genomes/"$2"/Mapping/SRA_Ids.txt"}' Genomic_accession_and_species_names.txt
than I just have to run the print an I get for each genome a file called SRA_Ids.txt.
From this file I can download the fastq files using the python script written by benjwoodcroft or the perl one written by Gerth & Hurst 2017.
But here is the point :
For each genome I will mapp the reads against it and get a coverage value for each scaffold usign Hisat2. But because I have a lot of genomes, if I download all the fastq available in biosample, it will take a lot of hard disk space. So I wondered if there was a way to add into the SRA_ids.txt file other informations, especially Total Bases (Mb) or at least the size of the R1/R2 files? The idea being that if the first two R1 and R2 fastq file already have a good amount of reads, I do not need to download and map the other fastq file.
Of course I could do it manually with https://ewels.github.io/sra-explorer/# for exemple but it would be time consuming so I wondered if there was not a fatest way to do it for all genomes.

Might be possible but you need a very customized approach for this. I guess it will be quite an effort to fully automate this. Why don't you split the entire dataset in like chunks of 10% (so download 10% of all files), map and evaluate if sufficient for your goal. If not, continue with the chunks until you are satisfied.
Log in to answer this question.
More posts like this
-
SRA download
written by sjlee4019 •
-
Tutorial: Setting up Aspera Connect (ascp) on Linux and macOS
written by ATpoint
-
Aspera Connect problem with "ascp: Taget address not available"
written by sbstevenlee •
-
EBI European Nucleotide Archive (ERA) aspera access broken
written by benformatics
-
Aspera: Failed to authenticate
written by tomas4482 •
-
Download problem of FASTQ files from the European Nucleotide Archive (ENA) using Aspera Connect
written by amzai •
-
ERROR: "ascp: Failed to open TCP connection for SSH, exiting."
written by dpc
-
Aspera ascp command line utility inside prefetch call to facilitate SRA data downloading
written by Denis
-
Downloading FASTQ with aspera
written by dagurim •
-
Download of FASTQ files from the European Nucleotide Archive (ENA) using Aspera
written by r.castellan •









Good work dude. Will use this next time I need to get data from ENA.
Thank you Sir, glad to help!
Good work! I once had the pleasure to use
fastq-dumpon whole-genome data. I was cursing in multiple languages! :DWhen I run the following commands on mac, I get this error message: ascp: Private key file not found at path /Users/antonellalisa/Applications/Aspera_Connect.app/Contents/Resources/asperaweb_id_dsa.openssh, exiting.
Session Stop (Error: Private key file not found at path /Users/antonellalisa/Applications/Aspera_Connect.app/Contents/Resources/asperaweb_id_dsa.openssh)
Can anyone help me with it please?
Hi mr.monahton,
sorry for that, on my Mac machine I manually renamed the
Aspera Connect.appfolder (which is the default name from the installer) toAspera_Connect.appbecause of the whitespace (which I hate). I updated the commands for Mac to work with the default installation path of the installer. Please try again with the updated command.I run into a problem when running the aspera installer on the linux cluster that I am using. The installer automatically tries to install in /bin and other root directories, but I don't have access to them, so I get an error. Is there a way to define a custom target directory for the installation? Thanks!
Hello and welcome to biostars lgbio ,
please open a new question for that. So we can keep the focus in this thread on the tutorial itself.
Thanks!
Can you confirm that you downloaded from the link I gave above (or at least the most recent version of the installer script) for linux? By default it installs to
~/.aspera/connect. Please double-check.I've recently started to download FASTQ files via Aspera, but I am using ena-file-downloader.jar. That's is the github link and it is also accessible from "Bulk Download Files" button in the same website. It has GUI, you can choose between FTP and Aspera and you can specify Aspera parameters. Do you think that the speed would be different between the applicaton and the terminal?
They probably both use the same Aspera server so speed is probably similar. Question would be if this tool you mention allows parallel downloads of several files.
I recently was downloading the data form CCLE experiment and it was taking ages (also crushing more than once) with
sra-toolkitandfastq-dump. I used your approach, slightly it modifying and it worked wonders! Thanks a lot!My modification below:
where
${tissue_of_interest}and${experiment}where variables I set up specifically to my needs (i.e. CERVIX, RNA-seq). I copied this intosra-explorer. The Project has too many files to directly searchsra-explorerwith its ID.Glad to hear it is used productively :)
I am sorry. I am considering switch from SRA because the files I want to download are too big and I got unkown error using SRA for some time. I am trying to use aspera client to download the fastq files. From the link above I downloaded the ibm-aspera-connect-3.9.9.177872-linux-g2.12-64.tar.gz and decompressed it to ibm-aspera-connect-3.9.9.177872-linux-g2.12-64.sh.
I also set the path to the directory I am working:
everything looks fine until the next step;
Which part did I made the mistake
PATH is the path to the folder containing the binaries, not the binary itself, please try:
PATH=$PATH:/home/caiqi/.aspera/connect/bin/Thank you for explanation. It is working and it is faster than I expected
Thank you very much for this useful tutorial. I followed the tutorial steps to download (GSE111653 dataset with BioSample accession number of PRJNA437670). First, I downloaded tarball file from Aspera client and then I ran tar zxvf /scratch/user/ye/ibm-aspera-connect-3.9.5.172984-linux-g2.12-64.tar.gz on linux.
Then, after downloading PRJNA437670.txt file from ENA, I ran the below command: $ awk 'FS="\t", OFS="\t" { gsub("ftp.sra.ebi.ac.uk", "era-fasp@fasp.sra.ebi.ac.uk:"); print }' /scratch/user/ye/PRJNA437670.txt | cut -f3 | awk -F ";" 'OFS="\n" {print $1, $2}' | awk NF | awk 'NR > 1, OFS="\n" {print "ascp -QT -l 300m -P33001 -i $HOME/.aspera/connect/etc/asperaweb_id_dsa.openssh" " " $1 " ."}' > download.txt
So, now, I have only 4 files in my /scratch/user/ye/ directory as follows:
download.txt ibm-aspera-connect-3.9.5.172984-linux-g2.12-64.sh ibm-aspera-connect-3.9.5.172984-linux-g2.12-64.tar.gz PRJNA437670.txt
I then ran the below command to download the data: $ cat /scratch/user/ye/download.txt | parallel "{}"
However, I faced with the following ERROR:
Academic tradition requires you to cite works you base your article on. When using programs that use GNU Parallel to process data for publication please cite: O. Tange (2011): GNU Parallel - The Command-Line Power Tool, ;login: The USENIX Magazine, February 2011:42-47.
This helps funding further development; AND IT WON'T COST YOU A CENT. If you pay 10000 EUR you should feel free to use GNU Parallel without citing.
To silence the citation notice: run 'parallel --citation'. Can't exec "/bin/sh": Argument list too long at /local/software/biobuilds/2017.11/bin/parallel line 3981. . . Can't exec "/bin/sh": Argument list too long at /local/software/biobuilds/2017.11/bin/parallel line 3981. /bin/bash: ascp: command not found /bin/bash: ascp: command not found . . /bin/bash: ascp: command not found Use of uninitialized value $opt::termseq in split at /local/software/biobuilds/2017.11/bin/parallel line 3608, <stdin> line 128.
Also, I tried:
$ while read LIST; do $LIST; done < /scratch/user/ye/download.txt
And I got many -bash: ascp: command not found messages
Would you please help me what I did wrong and how to fix it? Thank you very much.
Did you run the installer script for
Aspera? If not, do so.About the parallel command I cannot tell from here, maybe simply use a loop to download the files or install parallel again (or via
conda)Thanks for your reply. As you suggested, I ran the installer script for Aspera as below:
Then, I ran:
Then
$ while read LIST; do $LIST; done < /scratch/user/download.txtBut, again-bash: ascp: command not foundand again
cat /scratch/user/download.txt | parallel "{}"I would highly appreciate your help.
You can likely access the Aspera binary explicitly via
Maybe add this to your PATH variable, or modify the
awkcommand to include this full path.For
parallelto work, you will need to install GNU parallelYep, has to be in
PATHor call it explicitely as Kevin Blighe says. Same goes for parallel. If not done already, install it or use the loop. In either caseascphas to be inPATHor called explicitely.Thanks for your guide. I did as below, But, again -bash: ascp: command not found:
I can not see what I am doing wrong or missing. I really need your help to fix this problem. Many thanks.
I also wanted to add that I am using my university cluster which has a linux system. Many thanks.
Remove the
ascpfromexport PATH="/home/user/.aspera/connect/bin/ascp":$PATHit is onlyexport PATH=/home/user/.aspera/connect/bin/:$PATHalso without any quotation marks.The concept of PATH is that when entering a command (tool name) the system scans all folders in PATH for the presence of the called tool/executable. Therefore you only have to add the folder where
ascpis in, not the full path to the executable.Thank you so much for your great help. I removed ascp from export command as you suggested. I also used -i /home/fsgh1d18/.aspera/connect/etc/asperaweb_id_dsa.openssh" " " $1 " ."}' > download.txt in awk command. Now it is perfectly working. Many thanks.
Glad to help, if there are other issues feel free to ask :)
Many thanks for your support.
Thank you for this tutorial. I am stuck at this command line and got the following error message Any help would be appreciated.
command line:
error message
Hey, take a look at the comment trail, from here: C: Fast download of FASTQ files from the European Nucleotide Archive (ENA) Also, why are you using zsh?
It's a lot better than bash. Seriously, try it. It does everything bash does plus a lot more.
Used to be my preference back when I was running Open and Free BSD. Since then just used default
When used with OMZ, the fact that I can use any part of a file name for autocomplete (as opposed to having to use the first few characters on almost everything else) is a huge improvement for me.
Thank for the tutorial
Please do not add Answers, they do not serve any purpose on a Tutorial type post.
Has anyone noticed why, for the same run, FASTQ files downloaded from ENA are not the same size as those produced from SRA? Is there an obvious reason why that I might've missed?
My SRA procedure:
The fastq-dump options above are definitely recommended to get the kind of FASTQs from .sra files that are ready for mapping (see e.g. https://edwards.sdsu.edu/research/fastq-dump/).
If I gunzip those files and compare them to the gunzipped ENA ones they aren't same. Which source is the correct one?
Please ask a new question and delete this post. This post does not answer the top-level question and so does not belong as an answer.
Edit: Moved it to comment ;-)
From ENA:
From SRA with your command:
I don't see any difference. Can you be more specific what is "different" between them? Just use any of them. Don't overthink things. Just download the ENA one and start your alignment (or whatever you plan to do).
You counted lines, that's not going to tell you everything. The sizes are very different, and I thought maybe because ENA used higher compression but in my previous post the uncompressed are also very different in size.
And if you check md5 instead of lines:
I don't think it's overthinking to wonder why they aren't identical, since ENA is getting these data from SRA anyway, so aren't they doing the exact steps we just did and simply saving others time? Then they should've gotten the exact same result. Maybe they are doing some other processing of the files, would like to know what it might be.
Found the answer, which I suspected when I was writing back, that they modified the FASTQ entry headers:
Hi I'm installing your LoadDump script. I am finding that it will prefetch the *.sra files but parallel-fastq-dump is not processing them. Where is the --split-3 portion of the parallel-fastq-dump command on line 7 coming from? Thanks!
Please be more specific. What exactly is not working?
--split-3is a command fromfastq-dumpitself that splits paired-end reads into R1 and R2 (so two files in case the sra contains PE information). Usefastq-dump --helpfor more info.Thanks for your reply. I've copied the LoadDump script on your page into a file, made it an executable and am running it with a list of SRA IDs as you describe in your text. I have installed the sra toolkit, parallel, prefetch, parallel-fastq-dump and added their locations to my $PATH environment. While running the script I am finding that it is pulling down the .sra files and putting them into their own directories. However the script is generating the following messages: 2019-12-11T22:29:28 prefetch.2.10.0: 1) Downloading 'DRR168516'... 2019-12-11T22:29:28 prefetch.2.10.0: Downloading via https... 2019-12-11T22:29:33 prefetch.2.10.0: https download succeed 2019-12-11T22:29:33 prefetch.2.10.0: 1) 'DRR168516' was downloaded successfully [ERROR] DRR168516 apparently not successfully loaded
I am wondering where the error message is coming from and I was trying to understand if there was an executable called split-3 that was missing from my environment. Now I understand that this is coming from fastq-dump and I can see that this is in my $PATH.
Thanks for your help.
Please show the full command you used. It works fine on my machine. It seems that the same accession is being loaded multiple times.
I simply name my ID file IDs.txt and put it into the same directory as the script.
I just figured it out - it was a $PATH problem into the sra bin directory after all. All appears to be working fine now. Thank you very much for your excellent script!
Thanks for your reply. I've copied the LoadDump script on your page into a file, made it an executable and am running it with a list of SRA IDs as you describe in your text. I have installed the sra toolkit, parallel, prefetch, parallel-fastq-dump and added their locations to my $PATH environment. While running the script I am finding that it is pulling down the .sra files and putting them into their own directories. However the script is generating the following messages:
As I result I am seeing the SRA files are not being split by the function.
I am wondering where the error message is coming from and I was trying to understand if there was an executable called split-3 that was missing from my environment. Now I understand that this is coming from fastq-dump and I can see that this is in my $PATH.
Thanks for your help.
This is asking for my password yet I've never made a password. How can I get around this? EDIT: If you use this https://anaconda.org/rpetit3/aspera-connect it requires a password for some reason
I do not know. You can install it from the source that is linked which does not require root permissions. No need for
condahere.I finally got this to work. Do you know why I could be getting variable download speeds? The frist time I ran this, I was getting speeds of ~230 MB/s and now I'm getting 1.7 MB/s at best. Is there an option I can choose to increase the speed? What could be affecting this you think?
Could you clarify a few points for me please?
In your command line:
parallel-fastq-dump -s ${1}.sra -t 8 -O ./ --tmpdir ./ --split-3 --gzip && rm ${1}.sra
Are options
-s,-O,-s,--tmpdirrelated tofastq-dumpcommand or all of these belong toparallel-fastq-dump? I'm wondering, because i didn't find neither--tmpdirnor-scommand line options in my fastq-dump documentation (sratoolkit.2.10.0).SRAdata i was looking for. How i can check if all files were downloaded completely? Where i can find checksums for allfastqfiles i've downloaded?Yes, only
--split-3and--gzipare sratools options fromfastq-dump.parallel-fastq-dump -hexplains what the others are.In my experience when the download completed successfully, and only then, you will have a *.sra file on your disk. If anything goes wrong then
prefetchdoes not convert the temporary files it downloads to the final.sra. I never felt the need for checksums as this is pretty safe. I downloaded hundreds of files over the last years and never had any problem given that the download finished and I had a.srafile present.Thank you so much for the clarification! Unfortunately i downloaded several fastq files which for some reason i was not able to download via
prefetchdirectly from ENA website. How i can verify completeness of these fewfastqfiles?I think it is the same. ENA also writes files to a temporary file name unless everything is properly finished. If you get a final fastq file like
.fastq.gzit should be ok.Thank you again. Even if i used
wgetto download these? I have all the*.fastq.gzfiles in my output directory.Ah I see. Sorry I always download with
ascpand this uses tmp files. Withwgethmm, then you could indeed use the md5sums, I think these are available via ENA but I do not exactly know how, since I never used this. Sure you can find this in th documentation somewhere. Still, if the download did not indicate errors, you can also proceed right away and only if you run into trouble with corrupted files then go back and check integrity of the downloads.Which of the method is usually the fastest to download fastq files? I know it depends a lot on network yet over a stable connection (but low bandwidth) which one of wget, curl, aspera or SRA toolkit performs better?
Aspera will be the fastest , instead of curl or wget use Aria2 for a faster download.
SRAtoolkit downloads via http but it performs some internal validation checks and only will produce the final SRA file on disk if download finished successfully afaik, so for downloads from SRA I prefer it. NCBI does not support Aspera anymore.
For ENA it would be Aspera, this is why I cover it as the default method in this tutorial. Still, unless you download hundreds of GB or even TB standard download via wget is fine as well. With our HPC I tyically get download rates via wget from ENA in the ~50-60Mb/s range. Sufficient for most applications unless you are going to download entire cohorts.