Hi ! I'm currently a student and I have a hard time understanding some basics of bioinformatics; I'm currently learning about alignment, filtering, variant calling and such so my question might look silly but here it is anyway.
I have some trouble about how you work with paired-end sequencing files and what does it means to be paired-end.
After taking a look on the Internet I found an explanation of what is paired-end sequencing (tell me if I got it right):
For what I understood, a paired-end sequencing is just done by sequencing from A to Z and then from Z to A. Which will provide two distinct datasets, one for each direction.
My question is, when you are doing some alignment with tools like BWA, TopHat or whatever, do you have to reverse one of the two dataset or not ? Because, for instance, If I wanted to find a consensus sequence (or the position specific score matrices), if half of the data are in the wrong direction wouldn't it be completely wrong ?
Completely unrelated: I've also heard that TopHat should be used over BWA for aligning RNA, do you know why ?
 •
written
by
Sus •
•
written
by
Sus •

2 answers
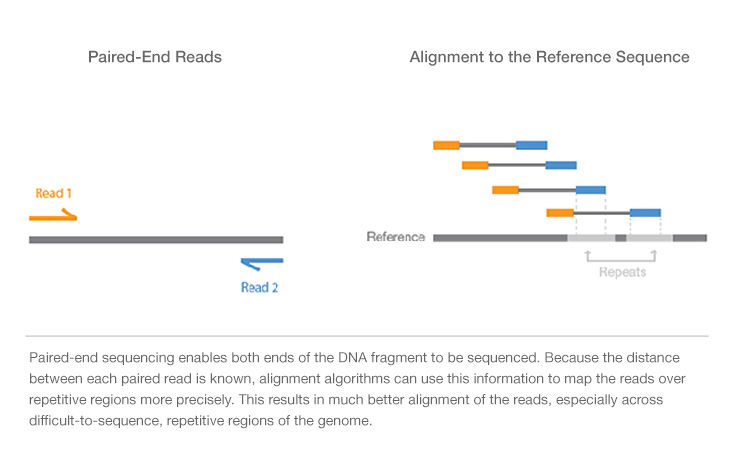
Always easier to illustrate with an image, from here. The grey represents the fragment, and each end of the fragment is sequenced. This allows more accurate mapping, particularly of repetitive regions. There's also a great animation here that illustrates the concept of Illumina paired end sequencing. As @h.mon stated, most programs will have parameters to deal with paired-end sequencing, and seriously, stay away from Tophat. STAR or HISAT2 are current alternatives
Most programs already take into account paired-end read orientation, you have to read the documentation carefully program-by-program.
Completely unrelated: I've also heard that TopHat should be used over BWA for aligning RNA, do you know why?
Don't use Tophat, there are several better programs, and it has been superseded by HISAT2 (from the same group of developers). BWA is not splice-aware, and Tophat is, hence Tophat is better for aligning RNAseq reads to a reference genome. But again, don't use Tophat.

Log in to answer this question.
-
How common is it to split fastq files prior to bwa mem to increase parallelization?
written by curious
-
BWA MEM -P option
written by chisqga •
-
paired end data with varying read length
written by manjumoorthy95 •
-
bwa: RNAME of chimeric reads in sam file
written by VHahaut
-
Mapping reads from Heterochromatin transcription in RNAseq
written by ghv8 •
-
How does BWA MEM handle repetitive regions on paired-end reads?
written by xianfan.jhu •
-
Orientation in paired-end sequencing?
written by John Smith
-
Paired-End Reads Alignment For Variant Calling ?
written by newDNASeqer
-
How To Generate A Paired End File Suitable For Bwa
written by GPR
-
About Paired-End Sequencing
written by Pierre Lindenbaum









