How to interpret a read hit with IGV?
Dear all,
I have aligned my sequences to a virus genome, I selected the sequence by IGV and BLAST it, obtaining a hit for Cytomegalovirus/HHV5:
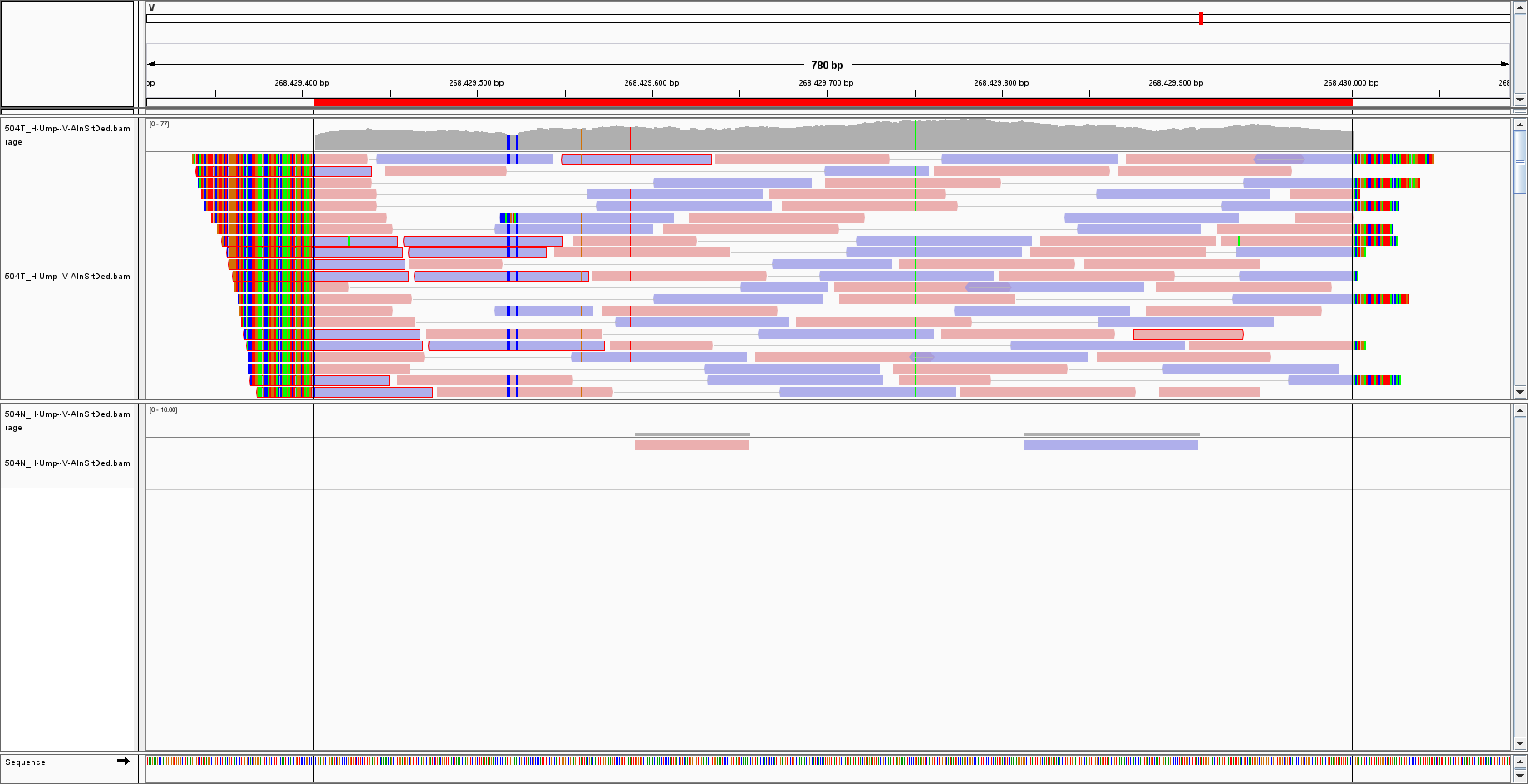 Now, the genome of CMV is over 200 000 bp, whereas the hit is around 700 bp. What happened to the rest of the genome?
Now, the genome of CMV is over 200 000 bp, whereas the hit is around 700 bp. What happened to the rest of the genome?
How shall I interpret the result? Could it be that there are few sequences of CMV in a vast majority of cellular DNA? (in that case, I would expect few reads but over the WHOLE CMV genome) Or is it simply a mapping error of the aligner? And how can I get a sure answer for the mapping then? The hit is present even with a mapping quality of 50, so it cannot be simply a spurious artefact.
Thank you
• 3,559 views
•
link
updated
by
Biostar
 •
written
by
marongiu.luigi
•
written
by
marongiu.luigi

•
written
by
marongiu.luigi
0 answers
No answers yet.
Log in to answer this question.
More posts like this
-
How to automate blasting contigs?
written by marongiu.luigi
-
Is there a model for detection of few reads for low abundance targets?
written by marongiu.luigi
-
Interpretation of sparse reads
written by marongiu.luigi
-
Application of empty blast result
written by marongiu.luigi
-
Hidden reads in IGV
written by marongiu.luigi
-
What sequence has being read by IGV's 'copy sequence'
written by marongiu.luigi
-
Identify insertions with IGV
written by marongiu.luigi
-
Sequence Length Distribution after trimmomatic
written by marongiu.luigi
-
fastqc report for degradome reads
written by Sam
-
how to calculate genome coverage for integrated genomes?
written by marongiu.luigi

Without sufficient information we can't interpret anything from this. Where is the sequence from? Is it known to be the genome you aligned against (presumably not)? Is the viral genome you're using related to CMV (most likely yes)? And so on...
I haven't understood the questions. "Where is the sequence from?", you mean from what kind of sample? human samples sequenced by WGS. I got the sequence by defining a region of interest in IGV and then selecting the 'copy sequence' option. "Is the viral genome you're using related to CMV?" the reference genome is a patchwork of some reference genomes including CMV.
You have WGS data from human samples, poi / then, you used BLAST to align these human sequences and found a 700bp hit for CMV?
It is possible to have CMV sequence in a human sample, depending on the origin of the human DNA sample and whether viral infection was present. It is also possible that your sequences align but have a very low alignment score. What were the results from BLAST?
Sorry for the delay, I had problems with IGV thus I could not retrieve the sequence. There are many hits on BLAST but are all HHV-5, just different strains listed. The mapping quality is set to 50 so I thought it was good enough. What should be a threshold to be sure of having good mapping results? And more in general, what is the procedure to determine whether a hit is a true mapping hit or just junk? Tx