I just do some practice on ballgown and stringtie, and I got some GTF file and ballgown`s file.
However, I just find I can't use R or something else to deliver a excel file which contain FPKM.
I think the excel file I want maybe looks like this:
gene_id FPKM
A 124
B 541
C 122
Please help me
Thanks a lot :)

2 answers
You can use standard unix commands for this as follows:
$ cat stringtie.txt
1 StringTie transcript 337772 338047 1000 + . gene_id "Zm00001d027250"; transcript_id "Zm00001d027250_T001"; cov "66.076088"; FPKM "8.407302"; TPM "14.141658";
1 StringTie exon 337772 338047 1000 + . gene_id "Zm00001d027250"; transcript_id "Zm00001d027250_T001"; exon_number "1"; cov "66.076088";
1 StringTie transcript 426764 432130 1000 + . gene_id "Zm00001d027254"; transcript_id "Zm00001d027254_T001"; cov "2.043083"; FPKM "0.259955"; TPM "0.437262";
1 StringTie exon 426764 426798 1000 + . gene_id "Zm00001d027254"; transcript_id "Zm00001d027254_T001"; exon_number "1"; cov "0.000000";
1 StringTie exon 426869 426970 1000 + . gene_id "Zm00001d027254"; transcript_id "Zm00001d027254_T001"
$ grep 'FPKM' temp.txt | cut -f9 | sed -r 's/gene_id "([^"]*).*FPKM "([^"]*).*/\1\t\2/g' | sed '1i#gene_id\tFPKM'
#gene_id FPKM
Zm00001d027250 8.407302
Zm00001d027254 0.259955

How can we get transcript level TPM values instead of gene level TPM values, I have tried to replace gene_id with transcript_id but it didn't work for me?

Assuming that your GTF file is same as above, you can do the following:
$ grep 'TPM' temp.txt | cut -f9 | sed -r 's/.*transcript_id "([^"]*).*TPM "([^"]*).*/\1\t\2/g' | sed '1i#transcript_id\tTPM'
Many thanks for your response, it work fine most of the lines till the pattern sustains like:
gene_id "MSTRG.26629"; transcript_id "AT5G53360.2"; cov "14.228090"; FPKM "5.268032"; TPM "8.616198";
But generates error when ref_gene_name in 9th column contains TPM letters in gene name (ATPMEPCRF)
gene_id "MSTRG.26631"; transcript_id "AT5G53370.1"; ref_gene_name "ATPMEPCRF"; cov "279.969208"; FPKM "103.660202"; TPM "169.542801";
Can you please check this issue?
 •
written
by
waqaskhokhar999
•
written
by
waqaskhokhar999

Form StringTie output you can use 'sample1_gene_abund.tab' file to extract these information,
FPKM:
cat {sample1}_gene_abund.tab | cut -d$'\t' -f1,8 | sed "s/FPKM/sample1_FPKM/" | sed 's/Gene ID/gene_id/' > {sample1}_gene_abund.fpkm.txt
TPM:
cat {sample1}_gene_abund.tab | cut -d$'\t' -f1,9 | sed "s/TPM/sample1_TPM/" | sed 's/Gene ID/gene_id/' > {sample1}_gene_abund.tpm.txt
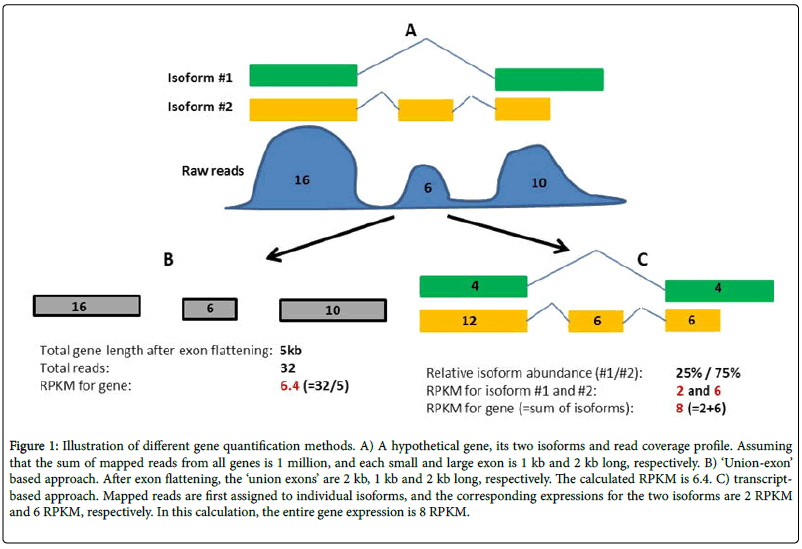
It is not always the case. There are also cases like this where it depends on the quantification approach,
Log in to answer this question.
-
Extraction of information from stringtie output for selected gene ids
written by kuntalasb •
-
How to get transcript ID and tpm from the gtf files of ballgown outputs?
written by newbie
-
Error when running Stringtie Merge: Error getting header ID# for gseq 1
written by nattzy94 •
-
Stringtie output gtf file only contains STRG,no original annotations
written by yiren •
-
print non match to list lines of GTF file
written by Sam
-
Add double qoutes around transcript_id in gtf file
written by BioBaby •
-
how to get counts/TPM/etc. from stringtie?
written by dec986
-
Generating FPKM matrix accross all samples after stringtie
written by ever_wudi •
-
stringtie (.gtf) vs ballgown (t_data.ctab) FPKM values do not match - why?
written by cpak1981
-
StringTie final sample gtf
written by samuel










Can you paste a few sample lines from the GTF file you are working with?
this is the top ten lines:
i think maybe a python script can extract the fpkm , but i dont know how to edit a complex python script.so i want to find some software to do this work.thanks a lot