Hi,
I'm working with a set of whole genome bisulfite sequencing data that has some quality issues I'm struggling to resolve satisfactorily. I was wondering if anyone has experienced something similiar and might be able to offer some advice.
It's HiSeq X, 150bp paired end reads, an insect species (and therefore very low methylation levels). After an initial alignment with Bismark I noticed that that the reported methylation levels would 'spike' at certain bases. Example (top = forward read, bottom = reverse):
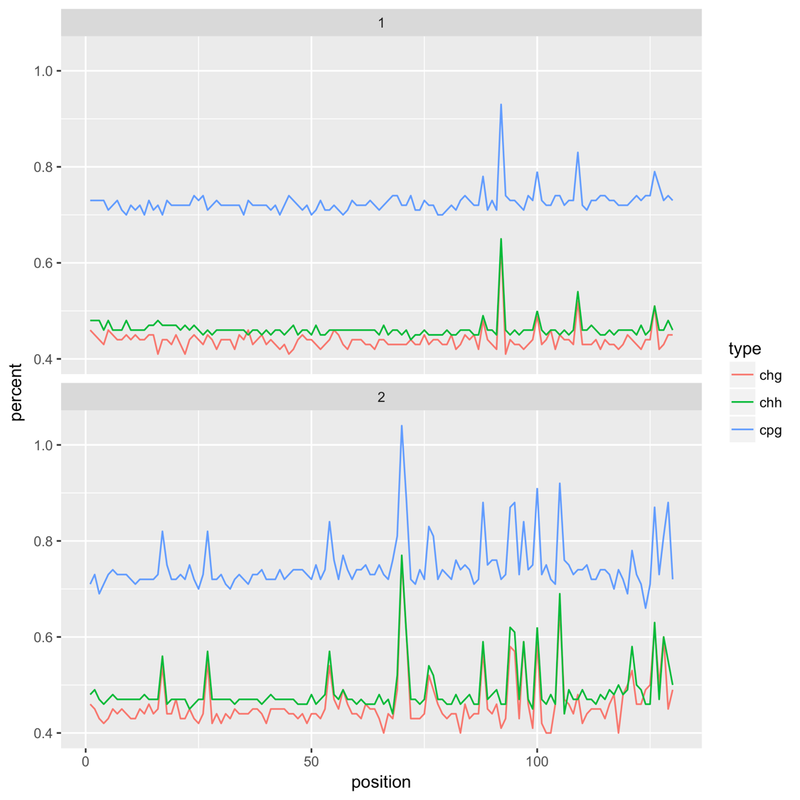
Checking back on the FastQC reports I found that this pattern went hand in hand with poor tile quality - the Per Tile Sequence Quality report would highlight tiles with issues at the positions where the spikes could be found. Extracting all the reads from a poor quality tile and running FASTX Toolkit's fastx_quality_stats on it generates a report that highlights the problem - the following is for the reverse reads of a bad tile, showing the per base sequence content. The same kind of issue can be found with forward reads, with C content spiking at certain bases:
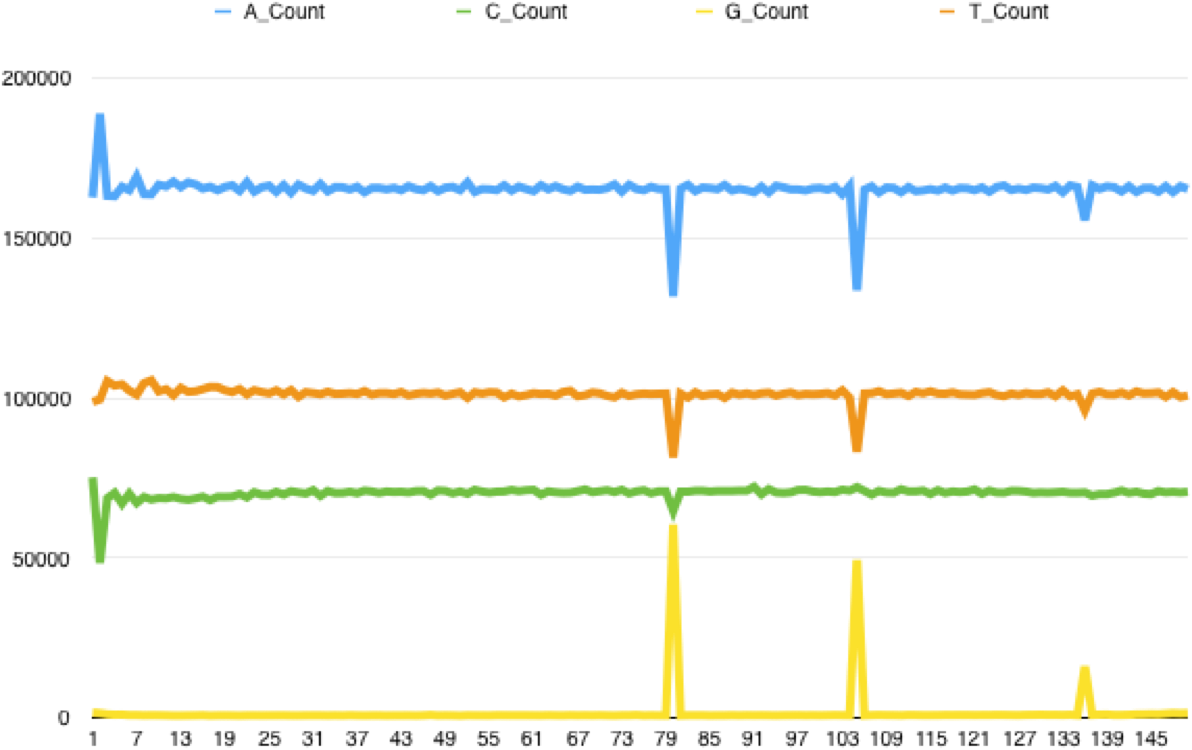
The tile pictured above is a particularly bad example; some tiles are less seriously affected with the percentage of C/G's called at certain positions rising only 2x above the read average. Looking at the reads themselves there is a pattern of sudden drops in base call quality along the length of the reads, affecting multiple reads at the same position. Overall it suggests to me that due to some instrument error the sequencer is calling a random base:

I'm using bbmap (specifically bbduk.sh) for data QC, but the usual quality filtering processes (qtrim, maq) don't solve it. I have tried using the filterbytile.sh function of bbmap to resolve this, but even using aggressive filtering this leaves the issue in place.
There are two solutions I've been looking at. The first is complete removal of affected tiles, but in some samples this results in the removal of nearly 50% of the data (although this isn't quite as bad as it first seems as after alignment and deduplication using Bismark the number of unique alignments reported is reduced by 20 - 25%)
The second is to separate out the bad tiles and subject them to extremely harsh QC, like so
bbduk.sh in1=badtiles.1.fq.gz in2=badtiles.2.fq.gz out1=badtilesqc.1.fq.gz out2=badtilesqc.2.fq.gz qtrim=rl trimq=40 overwrite=t minlen=20
When I tried this latter approach, however, it resulted in a drop in the reported methylation levels in the first 20 bases (see below). This is presumably the result of the harsh QC - if I had to guess it would be a number of much shortened reads with a compositional bias. This is something I'm still looking into, without understanding what's happened here I'm not keen on adopting it as a solution.
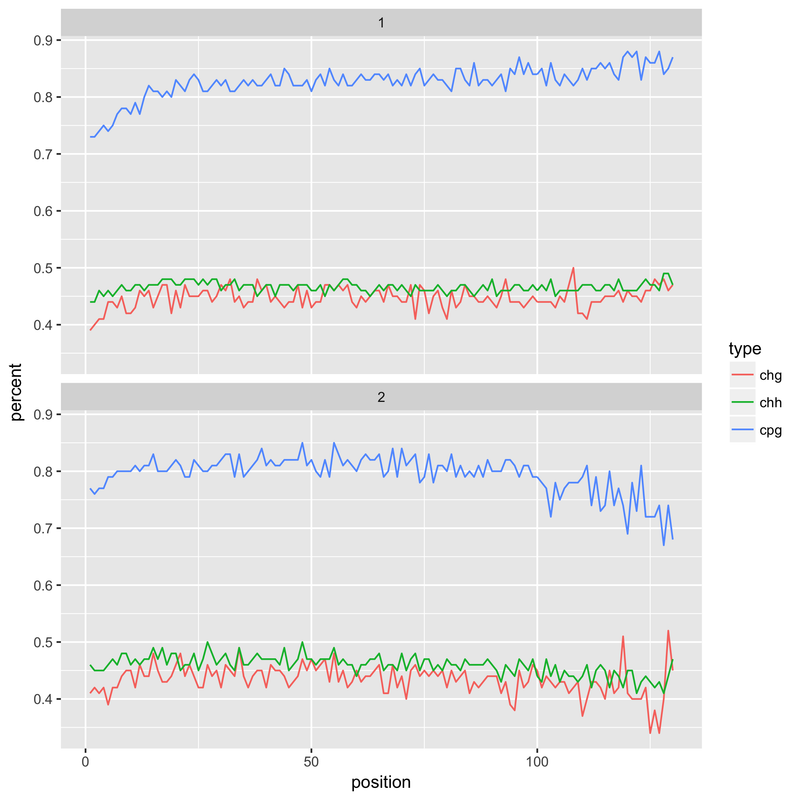
Any suggestions regarding this issue would be much appreciated!

0 answers
No answers yet.
Log in to answer this question.
-
FASTQC - Good "Per Base Sequence Quality" but Bad "Per Tile Sequence Quality"?
written by cg1440 •
-
Is it normal to obtain a perfectly dark blue "Per tile sequence quality" plot in fastqc?
written by cg1440 •
-
Filter per tile quality
written by gubrins
-
Any reason for higher reported levels of non-CG methylation when analysing non-directional WGBS lib…
written by benbio •
-
bad quality in certain tiles according to "Per tile sequence quality" in FASTQC
written by blur
-
Illumina HiSeq generates abnormally high duplication levels and per tile sequence quality patterns …
written by edikedikedikedik •
-
FastQC: per tile sequence quality after using filterbytile.sh
written by Nagesh •
-
BSMap to Bismark bam file formats
written by Jautis
-
per tile sequence quality decreases after QC with sickle
written by mary •
-
Why is sliding window applied to CpG methylation level calculation ?
written by hxlei613







