

finswimmer, thank you so much for the elaborate explanation. It really helps!
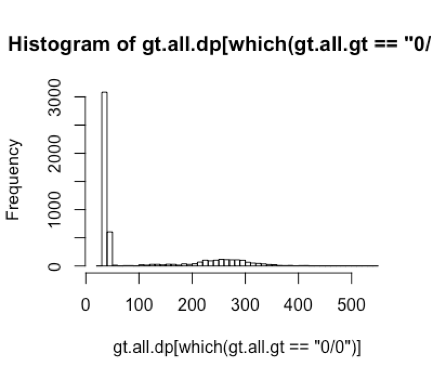
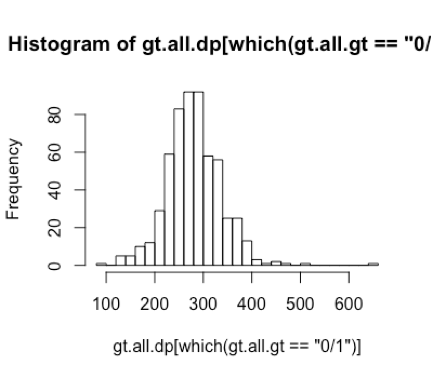
I think I understood my plot corretly and I was having problem expressing what I thought in English. Yes, I have a lot of samples with no variant at these positions. My samples are divided as cases and controls, and most case samples do not have variant here and most controls do. Assuming my samples are with the same ethnicity (which I checked), it is unlikely to find such a big difference in allele counts between case and controls with my sample size in a single gene. That's why I went to take a look at the variant quality. I was suspecting that maybe those 0/0s in the case samples are actually false negative calls since there's not enough read depth to align a 1 allele. Does this seem reasonable to you?
Could you tell me what do you mean by "your plot for genotype 0/0 should look more like in inversion of this with 0/1"? Thanks!
p.s. I further checked more gene regions and for some genes, two distributions (or three, containing 1/1) are similar. I guess this is a regional thing, i.e. some regions are "hard" to either sequence or call variant and they have more false negative calls










Hello,
Have a look at Kevin Blighe's post here: A: DP in VCF files?
fin swimmer
Thank you! I have read this post. According to Kevin's idea, it's safe to use a low DP cutoff (e.g. 10 or 18 for 0/1 and 1/1) to get accurate calls. But for 0/0, we may need higher values. I was wondering how can I decide which value to use...
Actually, when I thought about this again, it doens't matter what cutoff of DP to use for 0/0. Because if I filter the calls by DP of 40, for example, I get a ./. instead of 0/0. However, both of ./. and 0/0 are telling me the same thing: the genotype of this locus is unknown... :(
No, that is not strictly true.
0/0means that the variant caller was able to make a call at the site but that the call was homozygous reference../.indicates that there is not enough information such that a variant call could be made, and/or the evidence over the site has fallen below some threshold for QC.Thanks for the reply! I understand this is not strictly true. But according to the DP distributions in my data, those 0/0 calls with low DP seem to be false negative calls to me, i.e. some of them may be actually 0/1 but they are called 0/0 since there is no ALT reads aligned due to low coverage. In this case, there is no way to tell if they are "acutally" 0/0 or 0/1...
Yes, that's true. It's difficult for the variant callers, though, in part due to just how much error NGS reads can pick up. You could think about lowering your thresholds for:
...although, in doing this, you run the risk of picking up false-positives, as you have probably found.
Years ago, I just became so tired of this 'error' in NGS... it was around the time that my grey hairs first started to appear. I and a clinical genetics colleague of mine working together in a Children's Hospital in England came up with a new approach that pretty much solved this issue. After you obtain your final BAM prior to variant calling, do the following:
samtools mpileup)Kevin
Thank you so much for the explanation Kevin. I usually get VCF file from data provider so I haven't actually done variant calling myself. Many VCF files that I deal with have a lot of problems regarding variant quality and I do not always have access to the bam files. The only thing that I can do is to filter out "bad" variants as much as I could and keep as many variants in the same time. Do you, by any chance, have any suggestion on the filtering of VCF if such "error" already exists? Thanks again.
Not sure... can you not at least get the BAM files? The data provide should not have issues in sharing your own data with you (?). The VCFs are already heavily processed.
OK. I will ask about it. Thank you so much!
Hello again,
could you please tell something more about your graphics? Do they show the depth distribution for one variant in many samples? How does the plot looks like for 1/1?
fin swimmer
Hi, I made these two graphs using a few variants (5 to 6) within one gene called UBB (I also checked a few other genes). There is no 1/1 genotypes of these variants. The distributions are from these few variants in many samples (~1000 samples, all are EUR).