

Hmm, ok. Thank you for your answer! So I will not mess with the repaired bases at this point then and take care of it after creating the M-bias plot. I might have to use Bismark, since that's what is available on our cluster, and it has both the features that you mentioned above(3,4) . Is bwa-meth better for alignment than Bismark, in your experience?
Also, I just want to explicitly mention what I understood for the trimming part and confirm it. With Cutadapt, I should provide the reverse complement of adapter for the R2 file as well, right?
cutadapt -a AGATCGGAGAGC -A GCTCTCCGATCT -o oR1 -p oR2 inR1.fq inR2.fq
Cutadapt for paired end RRBS data
Hi all,
I have just started working with RRBS data, and I am having issue understanding the data and need to understand how to trim adapter sequences. This is probably a very naive question, but any help would be appreciated. I am trying to trim paired-end RRBS data using Cutadapt. (I am aware of Trim_galore, but I am using Cutadapt mainly because Cutadapt is already installed on the cluster that I am working on, and since Trim galore uses Cutadapt, I should get the same results).
If I want to trim the Illumina adapter sequence(AGATCGGAGAGC) from the 3' end of my Paired end data using cuadapt, at first I add 2 Ns to the Adapter (NNAGATCGGAGAGC) to remove bases added during end repair. So, my understanding is we are supposed to remove the additional bases and adapter only from 3'end. What happens to the adapters or additional bases at the 5'end? This paper, doi: 10.1186/s12864-016-2494-8, briefly mentions:
Removal of additional bases for pair-end sequencing can be tricky as it can affect subsequent RRBS read alignment. For example, removing two additional bases from the beginning of the read 2 (complementary reads to the original forward and reverse strands) would remove CGG tag that is used to search for indexed CCGG motif in the reference genome causing the reads to remain unaligned in RRBSMAP.
For my paired end reads, how should I trim the adapters using cutadapt? Do I not provide a reverse complement for my Read 2 at all ? or do it like below? Please advise.
cutadapt -a NNAGATCGGAGAGC -A GCTCTCCGATCTNN -o outputR1 -p outputR2 inputR1.fastq inputR2.fastq
Thanks!
1 answer
My suggestion is to not bother removing the repaired bases and to instead simply ignore them during methylation extraction. The procedure is then essentially:
cutadapt -a AGATCGGAGAGC ...(N.B., cutadapt can use multiple threads!)- Align (I usually use bwa-meth from Brent Pedersen)
- Look at the methylation bias (e.g.,
MethylDackel mbias ...) - Extract ignoring bases showing bias (e.g.,
MethylDackel extract --OT 2,98,2,98 ...)
Then you're maximizing the quality of the alignments but also not including problematic bases (or having your cake and eating it too, if you prefer).
• 1 views
•
link
written
by
Devon Ryan

- I've typically gotten better and faster results with bwa-meth, yes.
- You can use
cutadapt -a AGATCGGAAGAGC -A AGATCGGAAGAGC, you don't need to reverse complement the adapter for R2 since the read will have the opposite orientation to begin with.
• 1 views
•
link
written
by
Devon Ryan
Hi Devon, Just a follow up on the last question. I trimmed the files and ran bismark with default options as below:
- Genome preparation:
bismark_genome_preparation --verbose hg38_gencode - Alignment:
bismark reference_genome -1 Read_1.fq -2 Read_2.fq
The M-bias plots look like these (below). In the M-bias plot examples (Google), the CpG plot line looks like a straight line across the length of the read except at ends. Mine are very uneven across the read length. Would you know why that is?
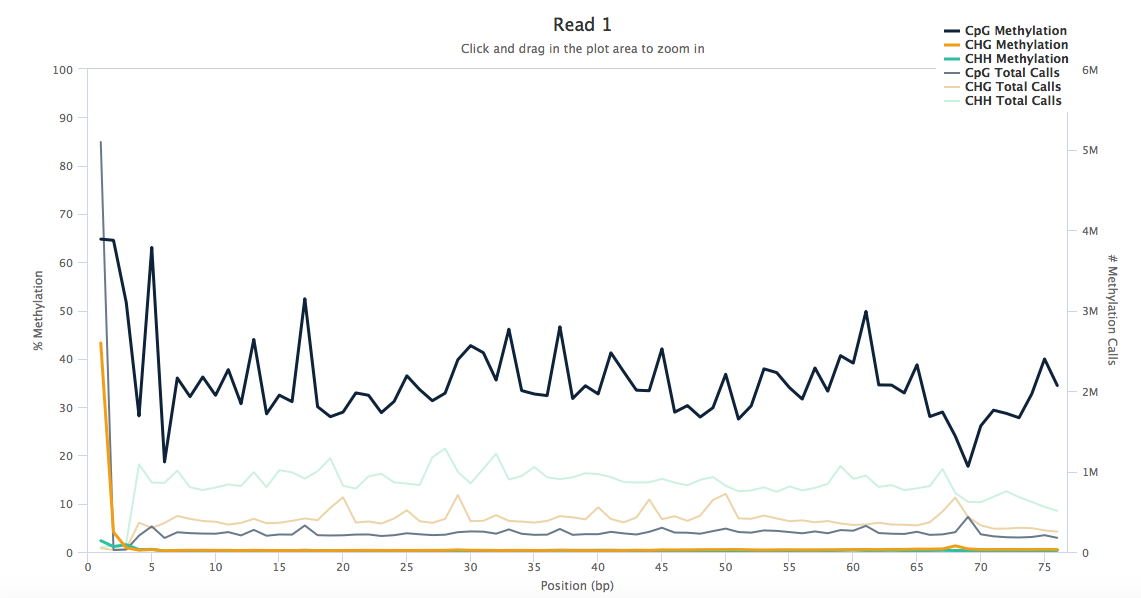
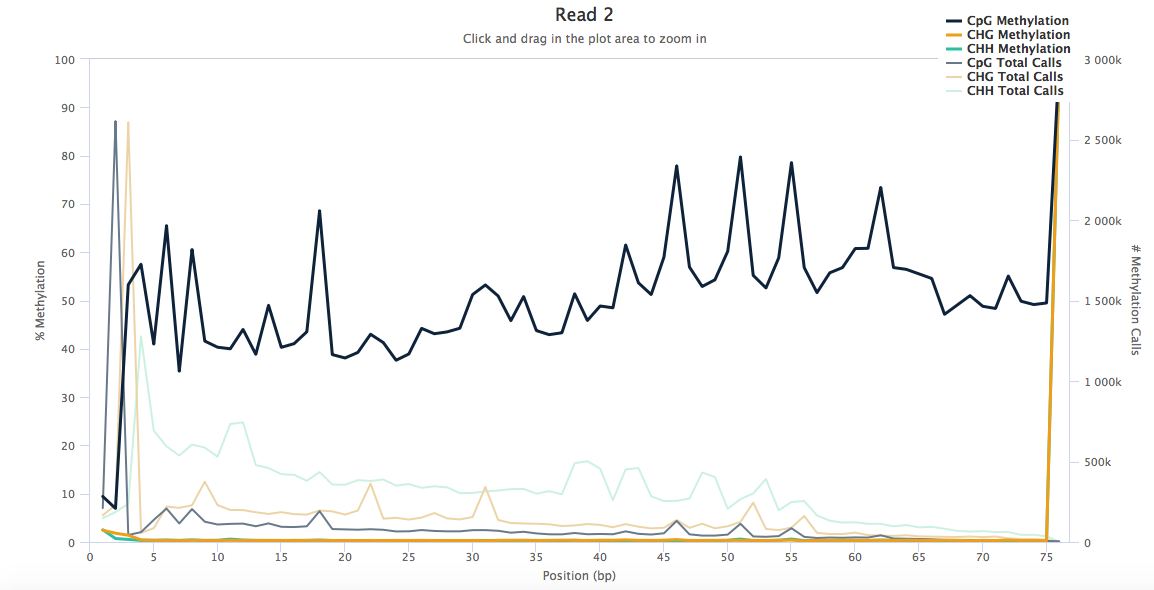
Yikes! What was your alignment rate? I usually only see M-bias plots like this with targeted amplicon-seq combined with bisulfite treatment (i.e., really really reduced representation BS-seq). Do you have a high degree of duplication? That's the most likely cause of something like this. If that's the case, then mark duplicates and recreate the M-bias plots while ignoring marked duplicates (I presume there's an option for that in bismark, but if not just use MethylDackel).
• 1 views
•
link
written
by
Devon Ryan
Log in to answer this question.
More posts like this
-
Using Cutadapt to trim adapters from paired-end small RNA sequence data
written by melissa.joubert •
-
How to remove Illumina TruSeq Index adapters reverse (R2 reads )
written by Info.shi •
-
Cutadapt or Trim Galore for SMART-seq?
written by Youyy •
-
ATAC-Seq kmer removal
written by elenichri •
-
Cutadapt wrong behaviour
written by dllopezr •
-
Does Cutadapt change the x,y coordinates in the fastq read name?
written by drkennetz
-
Adapter Removal regardless posistion
written by biofalconch
-
How can I remove adapter sequence from illumina 2000 paired end data?
written by tcf.hcdg
-
cutadapt: forward and reverse adapter sequence options
written by umn_bist •
-
trim_galore and cutadapt to paired-end DNA sequencing
written by Shicheng Guo









