Hello everyone,
I'm pretty new to the field, so forgive me if this is a dumb question. So, I am working on mapping out single reads to the reference mouse genome, with given 260 fastq files. What I'd like to do is make all 260 files to be good quality reads. Right now it's a total mess, as in some have bad 'per base sequence quality' and 'per base sequence content', while some reads have bad 'per base GC content', and so on.
My original thought was run a program that fetches what passed and what failed from summary.txt from each fastqc directory. Then I'd have list of files that failed 'per base sequence content' or failed 'per base GC content' and such. However, because each reads are different within the list, I would still have to open html file and look individually to improve the quality. ( ex. I might have to trim 10 bp while other files need to trim more than that to improve the quality. )
Is there a way to somehow program it so that I can make all the files to be good quality reads at once, rather than checking each fastqc.html files myself and make different adjustments to each reads to improve the quality?


3 answers
Try multiqc, it collects all the fastqc reports into one overall report - extremely handy if you have lots of files.

I would ask to take a look at AfterQC . It can walk through all FASTQ files together in one folder and produce all sorts of QC plots in different sub-category folders.
Not each report but multi-report will produce a plot like 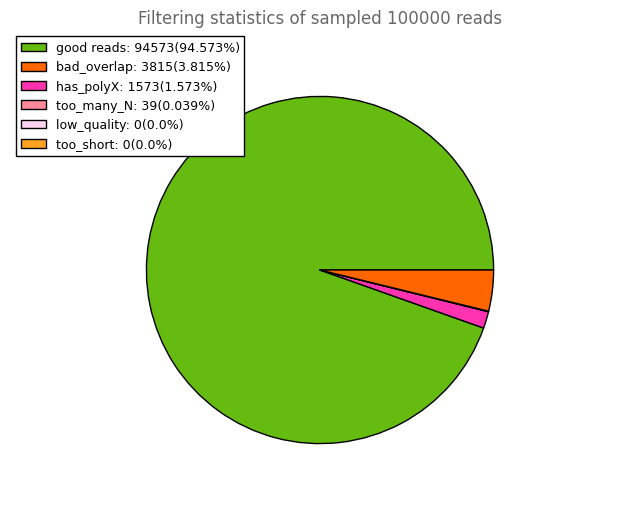 and other subsidiary plots for all the samples for your better understanding. This should be helpful.
and other subsidiary plots for all the samples for your better understanding. This should be helpful.

Don't worry, I don't think it's a dumb question. QC is important but just screening a bunch of htmls will make you crazy. I'm aware of a tool which enables you to perform QC in tabular format, but can't remember the name. Someone else will come along and help us both I guess.
But with regard to your solution, in which you propose to improve the quality per sample by trimming 10bp or more or less I really have to disagree. This is not the way to go. You have to treat all your samples equally and perform processing in a similar fashion, arbitrary making a judgment on treatment will create a huge bias and invalid results. Stick to one strategy. Perhaps optimize on a few samples. You also shouldn't look only to the effect on the (possibly flawed) fastQC plots, but test the effect of your trimming on the mapping metrics. (Exaggeration: trimming your reads to high quality reads of 5 nucleotides might have a nice effect on quality, but mapping will be a disaster.)
Hopefully your samples are not that heterogeneous that a common trimming strategy doesn't work for some, because that's already bad news for any downstream analysis. This would seem like a serious batch effect.

Log in to answer this question.
-
Fastp Trimming for WGS
written by j.k3096 •
-
Trimmomatic - To trim the low quality reads
written by sainavyav22 •
-
Read length for single -ended RNA seq data analysis
written by vinaya •
-
Bad Per sequence GC content
written by Ekaterina D •
-
how to remove warning and failures observed inQuality report by Fastqc for RNA-Seq data
written by aiswaryabioinfo •
-
FastQC: per tile sequence quality after using filterbytile.sh
written by Nagesh •
-
Interpretation of FASTQC results - do I need to trim my sequence?
written by sallyanndunn18 •
-
Questions regarding proprocess for raw data and usage of FastQC
written by mangfu100
-
Ngs-Qc-Per Base Sequence Content Failed
written by williamjohn360
-
About The Priority Of Trimming
written by Tonyzeng •










+1 for MultiQC mentioned below. It can handle logs from several other applications (besides FastQC like aligners etc) and consolidate them in one location.