


Thank you for your reply @genomax. However, I did not understand your statement "Two reads coming from the fragment being sequenced would align much farther apart than just the size of the insert, if the fragment contains a splice site". Still, I am a bit confused about when to use single and paired-end sequencing?
Forum: Single-end sequencing versus paired-end
1 answer
I will refer you to following diagram that should explain how paired-end reads provide information about how long the fragment is that is being sequenced when a reference is available for mapping (Image comes from A: What is the different between Read and Fragment in RNA-seq? ). Two reads coming from the fragment being sequenced would align much farther apart than just the size of the insert, if the fragment contains a splice site.
Single-end reads would be equivalent to Read 1 or Read 2 on its own without the mate.
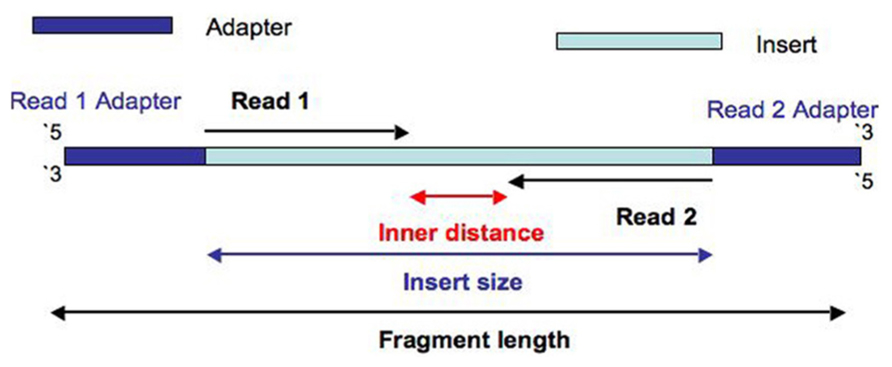
See image below. Red or gray bars connected with dotted lines represent two ends of fragments that originated from exons being spliced together (don't worry about different types of splicing depicted). Paired-end reads sequence the two ends of such fragments. By aligning the data to the reference one will be able to figure out that those two reads came from two exons that are a certain distance apart in the genome (> size of the fragment sequenced). If you are interested in this type of information (even if you don't have a reference, since you can do de novo transcriptome assembly with such data) they you will want to do paired-end sequencing. If you were interested in long scale genome rearrangements (e.g. translocations in cancer etc) then you will want to do paired-end sequencing (though no splicing will be involved here).
(source: https://www.frontiersin.org/articles/10.3389/fgene.2019.01152/ ).
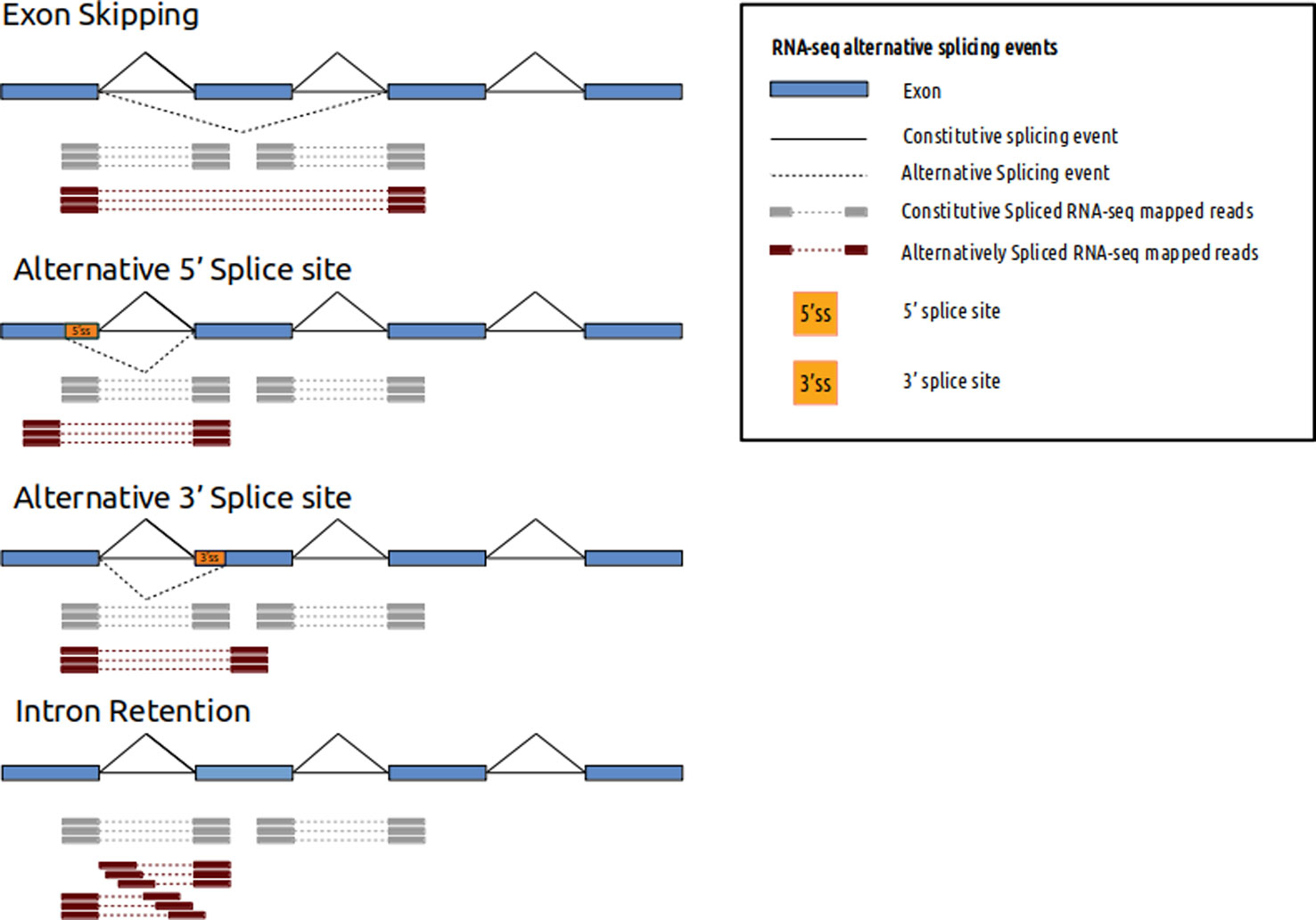
If you have tight budgets then you could save some money by doing single-end sequencing for pure counting applications. For applications like ChIPseq you can do single end sequencing since you are not really looking for information w.r.t the reference at a longer distance.
Log in to answer this question.
More posts like this
-
What should be control group for differential expression in single cell? either distant normal samp…
written by gorizwango •
-
The use of RNA-Seq to analyse expression
written by Anonymous •
-
Validation tools in bioinformatics
written by tonmoya.22 •
-
Whole genome sequencing or whole exome sequencing- which is better and why?
written by Sujata •
-
Polymorphic sites detection from MiSeq data of 3 individuals
written by sujata •
-
variant calling using pseudoaligners
written by jmgoldstein7 •
-
converting paired end reads to single end, is this a good idea?
written by John
-
A question about differential expression analysis
written by tunl
-
Interpretation Of Differential Exon Usage Results
written by Sam
-
Tutorial: Introduction To Ngs Bioinformatics By The Bioinformatics Team At The University Of Texas
written by Istvan Albert









Single versus Paired-end
So, if a single-end reads spans across two exons, will it not be possible to figure out the same here?
It should be possible to do that. Only difference is here you are going to be looking at say 50-100 bp read. Where as with a paired-end data you are sampling a 350-450 bp fragment.