Hello,
I am using Seurat package for the analysis of my single-cell RNA-seq. I read their basic PBMCs tutorial https://satijalab.org/seurat/v3.1/pbmc3k_tutorial.html. However, I get confused about some steps. I would highly appreciate having your answers on my following questions:
1) Is FindVariableFeatures() working on Normalized-Data or on Scaled_Data?
As in the seurat tutorial it is after NormalizeData() but before ScaleData(), but in kallisto | bustools tutorial (file://filestore.soton.ac.uk/users/fsgh1d18/mydesktop/Kallisto_bustools/BUS_notebooks_R-master/docs/10xv2.html), it is after both NormalizeData() and ScaleData().
2) Is FindMarkers() working on Normalized_Data or Scaled_Data or clusters obtained by RunTSNE()/RunUMAP()?
As FindMarkers() finds marker genes of the clusters, so I assume that the input data for FindMarkers() should be clusters obtained by RunTSNE()/RunUMAP() which these functions are also using scaled_data or selected_PCAs (obtained from RunPCA function), see below. Therefore, in practice, FindMarkers() is also using Scaled_Data, while it should work on Normalized_Data.
RunPCA() on Scaled_Data --> Selection of PCAs --> RunTSNE()/RunUMAP() --> FindMarkers()
3) If FindMarkers() works on Normalized_Data (rather than Scaled_Data), how it can be explained that FindMarkers() has inputs of clusters obtained by RunTSNE()/RunUMAP() which these functions are also using scaled_data (not Normalized_Data)?
Many thanks for any help and clarifications.


1 answer
You appear to have a few misconceptions about what Seurat is doing for each command. I will do my best to address them.
Neither, it's just using the counts. This step is just identifying the most variables genes to help limit the non-sparse matrix returned by
ScaleData, as it is very memory intensive and will bloat the object greatly if all genes are used. In addition, the top few thousand most variable genes have been shown to be plenty sufficient for marker identification.RunTSNEandRunUMAPdo not perform clustering, that is performed byFindClusters.FindMarkersuses thedataslot by default (normalized counts) - it would make no sense to use thescaled.dataslot, as those are basically just z-scores (or residuals if usingSCTransformor one of the integration methods). Depending on the test used, it may make more sense to use thecountsslot at times.Again, these methods are not performing clustering. Clustering is performed by
FindClustersafter constructing a shared nearest neighbor graph on the output ofRunPCAviaFindNeighbors, which uses the PCA embeddings to determine similarities between cells. The clustering function then groups cells based on these similarities into clusters with an adjustable resolution that defines how granular the distinctions between clusters should be.
I highly recommend reading the manual for the commands in question to better understand what they're doing.



Hi Jared.
Just one more about the second point of your answer.
it would make no sense to use the
scaled.dataslot, as those are basically just z-scores (or residuals if usingSCTransformor one of the integration methods). Depending on the test used, it may make more sense to use thecountsslot at times.
What I understand was SCTransform will only calculate residuals and we can access it by GetAssayData(obj, slot="scale.data"). If I understand you correctly, the value of GetAssayData(obj, slot="data") is also calculated by SCTransform and such value is done by NormalizeData() in old Seurat. So is SCTransform's GetAssayData(obj, slot="data") == NormalizeData(obj)?
Since Seurat is under development continuously and there is always an 'A-ha' monent.

I do not know. I expect they are similar, but I don't know enough about the internals of the SCTranscform function to say that they should be identical. Easy enough to test yourself on a toy dataset though.
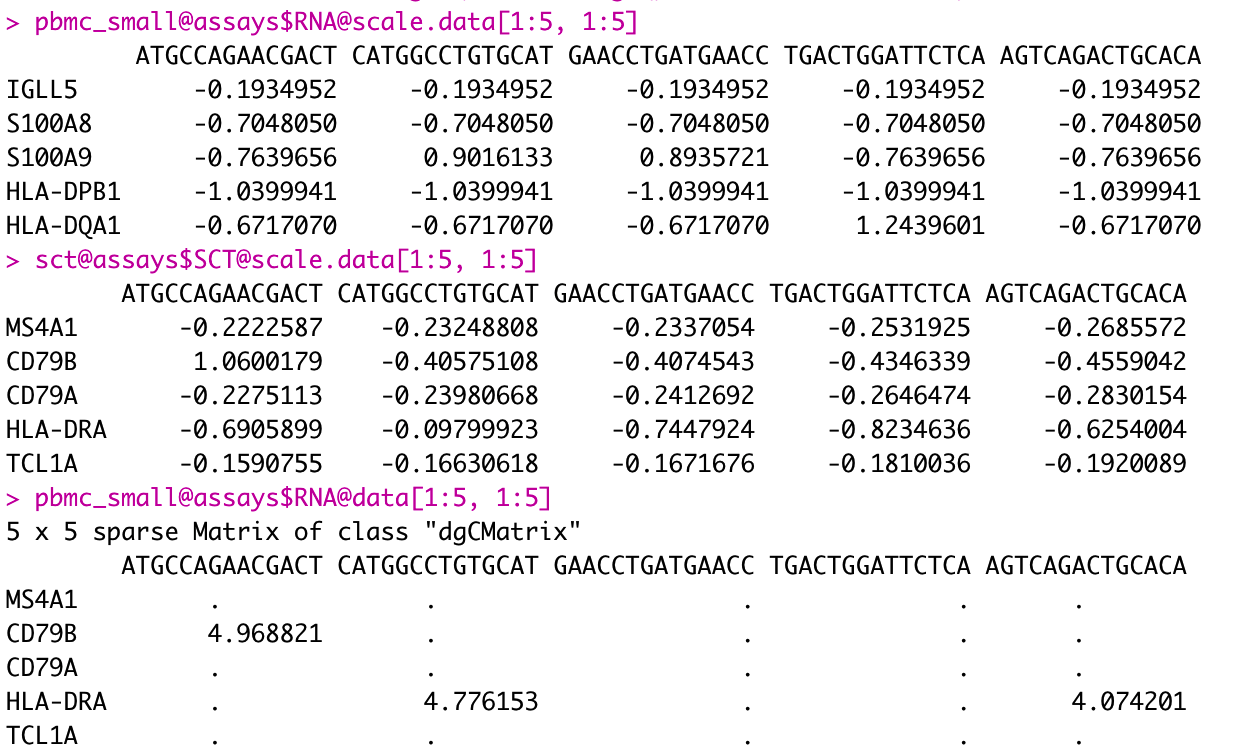
Just double-checked.
RNA
counts: Stores unnormalized data such as raw counts or TPMsdata: Normalized data matrixscale.data: Scaled data matrix
SCT
counts: corrected countsdata: log1p(counts)scale.data: pearson residuals
So SCTransform's GetAssayData(obj, slot="data") is not equal to RNA's NormalizeData(obj).
But another big issue I can not make clear is when we should which assay or slot even I spent more than 3 hours on this. Especially for FindMarkers.
I am not sure if this will help but this is my current understanding. I have recently started learning about the Single Cell analysis and here it is.

They contradict point 2 in numerous other issues. It has been nearly 2 years since they introduced that function and it's still not clear how/if SCT normalized values are truly appropriate for DE in all cases or not.
The answer to "can I use the SCT assay with typical DE function available in Seurat?" from the Satija lab seems to be something along the lines of "Maybe. Probably. Sometimes. Sometimes probably not."
On the other hand, the sctransform author (the package, not the Seurat SCTransform function) has provided a way to do DE on sctransform'ed matrices (including Seurat SCT assays), so who knows at this point. See the vignette.
Log in to answer this question.
-
Finding high number of significant genes by FindMarkers by Seurat
written by Rosa Icela •
-
Seurat object with multiple PCAs/UMAPs from different assays
written by solo.albif •
-
sci-RNA-seq
written by kilcdincer •
-
SIngle cell analysis
written by JY •
-
Subclustering of intergated cells from scRNA-seq data
written by fifty_fifty •
-
Trajectory analysis using Monocle3 with Seurat sub-clustering
written by joonhong kwon •
-
How to run UMAP on single cell results from 2 different runs?
written by bioinfo •
-
Seurat CellCycleScoring – confused about the proper order of operations when using SCTransform
written by GPM •
-
Using SCTransform with Seurat for multi-sample RNA-seq data
written by steveh •
-
scRNA-seq, SEURAT, NormalizeData, ScaleData, PCA, CCA ...
written by Bogdan









