
I want to make a pipeline that can take sam files from different aligners like HISAT2. One thing I like about HISAT2 is you can either map the reads to a transcripts or a GTF file if you had one available. What I like about BBMAP is that it has minid:
minid=0.76 Approximate minimum alignment identity to look for. Higher is faster and less sensitive.
CLC (a proprietary tool) has similarity and lengthfraction but I'm wondering if there are any downstream tools that can do this from the sam/bam file that is also computationally efficient. If I wrote something in Python it would take a long time (plus I don't really know what I'm doing for this type of work...I deal mostly with downstream data)
-s --similarity Set similarity score (default 0.8).
-l --lengthfraction Set length fraction (default 0.5).
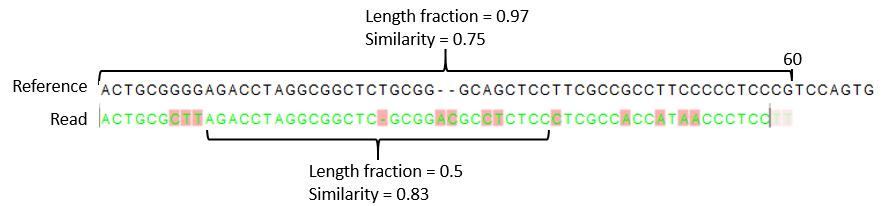 Is there a tool that takes in
Is there a tool that takes in sam/bam as input and take parameters like similarity and lengthfraction(like CLC) that outputs a filtered sam/bam file?
I don't know any tool that can do that automatically.
I guess the easiest way is to parse each alignment with the softclips in the CIGAR string for the length fraction and the MD-tag for the similarity. If your BAM file is encoded in sam1.4, you can just use the CIGAR string.
If you want to compare different alignments, you can also include the BAM files' mapping quality.