Hi,
I am working on fungal isolates from deep-sea sediments. It is debated on whether these isolates are haploid or diploid. I was wondering if there was a way to determine if they were either haploid or diploid using some type of bioinformatics tool.
I did try assembling the genome using both SPAdes and diploid SPAdes. Diploid SPAdes seems to be the better assembly, but I do not think this means I can ultimately say that these fungi are diploid.
Thanks in advance for your help!
 •
written
by
Morgan S. •
•
written
by
Morgan S. •

2 answers
You might try to analyze the kmer distribution from your reads. For this, you might use
- KmerGenie: http://kmergenie.bx.psu.edu/ , or
- Jellyfish: http://www.cbcb.umd.edu/software/jellyfish/
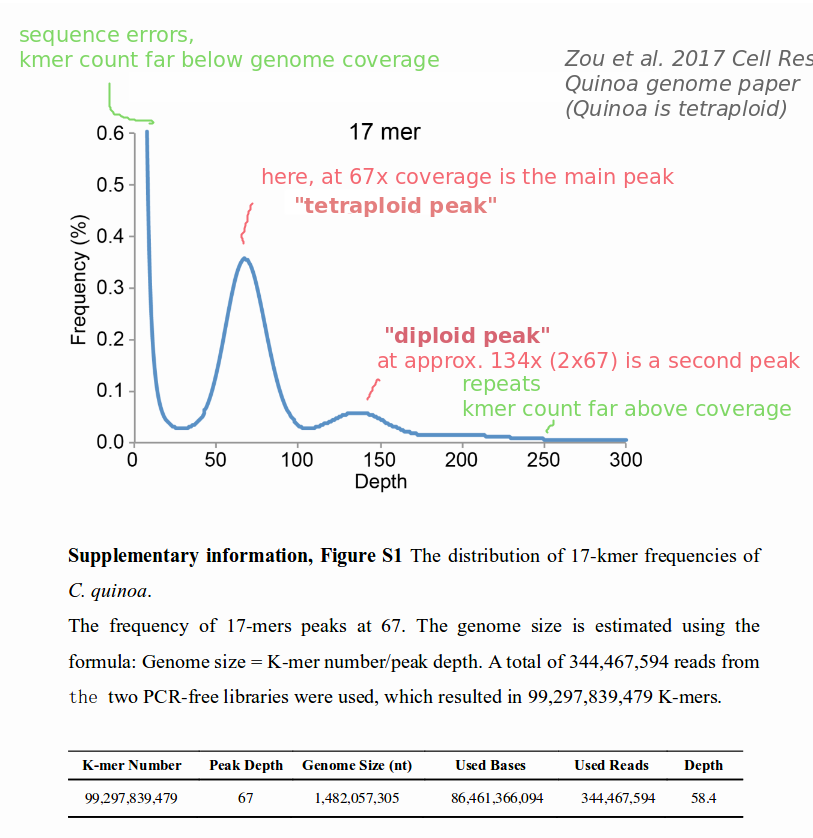
and count your kmers for a few different kmer lengths, e.g. 17mers as in the example.
Then, you would need to plot your data as in the figure below. X-axis: kmer depth, how often each kmer is counted, Y-axis: frequency, how many individual kmers are counted with this kmer depth.
The example is taken from the quinoa genome sequence by Zou et al. (2017). Quinoa is tetraploid. The graph is typical for a tetraploid organism as it has two peaks, a diploid and a tetraploid peak. A diploid organism would only have one peak,
You should check what it looks like for your species. Best luck!

Given that you have assemblies already and provided your strains are heterozygous wild strains (and not homozygous lab "wildtypes"), you can map the reads back to your assembly and predict variants on the assemblies. In haploids, you'd expect very few variants, in diploids many heterozygous mutations with an allele frequency distribution close to 50%. You can read this information directly in the vcf file from the variant caller and process it in Excel.
Using this, you can resolve up to tetraploidy, potentially even hexaploidy. I can't find the reference, though...
Probably not less work than toheitka's solution, but a different approach.
Log in to answer this question.
-
metagenome binning with MetaBAT2, and Maxbin2 created bins for some samples but not others.
written by Irshad •
-
Suggestions for Alternate Assembly Resources to Trinity
written by kacollier •
-
Normalization of raw Illumina reads.
written by robert.murphy •
-
Diploid genome gene annotation
written by Morgan S. •
-
Genome Guided Transcriptome assembly
written by Morgan S. •
-
SPAdes output with assembling warning
written by DanielC
-
To reassemble Illumina and PacBio, or just upgrade previous assembly with PacBio?
written by Morgan S. •
-
MAKER annotation pipeline output
written by Morgan S. •
-
reference-guided genome assembly: stretches of Ns
written by User 4014 •
-
Assembly method validation - am I doing it right?
written by f2369583 •






You can try
kmercountexact.shto test for ploidy (http://seqanswers.com/forums/showthread.php?t=64086 ).Additional link that is useful.