When I first wrote a duplicate-removal program, I was worried about the possible presence of "sister" duplicates, so I designed it to detect both identical and reverse-complementary duplicates. I was not concerned about the presence of these duplicates because DNA is double-stranded and thus a fragment might spawn two clusters - in fact, I doubt that's likely. I assume that when DNA is fragmented, the two strands will generally break independently. Rather, I was concerned that during PCR, a single fragment might spawn both identical and reverse-complementary fragments, so both would need to be removed.
However, in testing, I found that the rate of identical fragments was high, and the rate of reverse-complementary fragments was extremely low - a rate that could be completely accounted for by coincidence. So, I don't worry about it any more. Though I should mention that I don't really understand why this is the case - for a PCR-amplified library it still seems to me that reverse-complementary duplicates should be present. Perhaps someone else has some insight here?
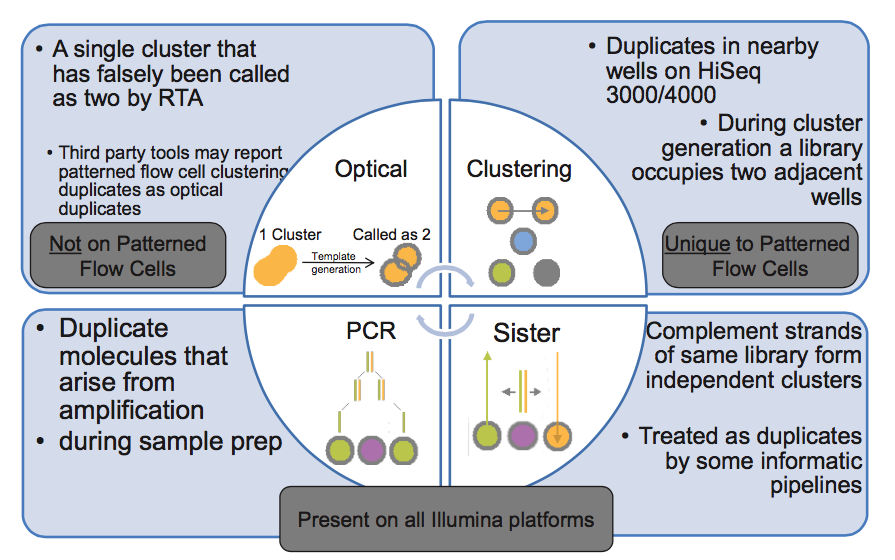





















Does not it mean, that if you have double stranded (complement strands), when each strand is create independent cluster position. So after sequencing you have the same data set but in reverse/complement orientations, but after alignment to reference is the same starting position = flag as duplicates?
Well I don't think something like Picard markduplicates would mark it as a duplicate if sequence is the same but the strand flag is different. Need to check it to be sure.
Correct - reads that map to the same position but on opposite strands are not marked as duplicates by Picard, nor are they removed by SAMtools rmdup command. But those reads would have to be from independent library molecules (i.e., not duplicates). Read one always proceeds from the same adapter end (I believe it's P5) so, even if sister clusters were formed from the two strands of a single library molecule, their orientations relative to P5 would be identical.
I don't get it. You would have P5 on one strand and P5's complement on the other, would you not? And the complement should not be able to attach to the flow cell.
You're conflating two issues. My point was that, if two reads have opposite orientations, they cannot be duplicates of any type (PCR, sister, or optical) b/c all inserts are sequenced relative to the same adapter end and strand.
Regarding annealing, the flow cell contains both P5 and P7 anchor primers. These serve as PCR primers for bridge amplification (aka clustering). If one strand of the library anneals to P5 (i.e., is the reverse complement), then the other end of the other strand MUST be the reverse complement of P7 (draw a diagram if this is unclear). But one of the two anchor primers is blocked during annealing (see discussion below), which is why sister duplicates are not produced.
Got it, thank you for the explanation.
A blog post has appeared discussing HiSeq 4000 duplicates from Babraham group: https://sequencing.qcfail.com/articles/illumina-patterned-flow-cells-generate-duplicated-sequences
This has also been discussed extensively in this SeqAnswers thread: http://seqanswers.com/forums/showthread.php?t=72901