Greetings!
I want to check whether my BAM file suffers from GC-bias, especially because this is a relatively very old sample, with the following details:
- Tissue Extraction Kit = GeneAll HybridR+ kit (GeneAll)
- RNA Extraction Kit = TruSeq RNA sample preparation kit
- Platform = Illumina HiSeq 2000
- Sequencing Kit = TruSeq SBS kit v3-HS
Based on several posts here on BioStars and elsewhere online, I chose to use
I've generated plots for 4 different combos:
- before vs after correcting for GC-bias,
- using either the full unaltered genome assembly or the repeat masked genome assembly.
I seek your help with making sure my interpretation of these results are correct, so here is a composite image of the plots.
If you can confirm / refute my statements, and answer my questions, with any relevant links and commentary, I'd be most obliged. Thanks in advance!
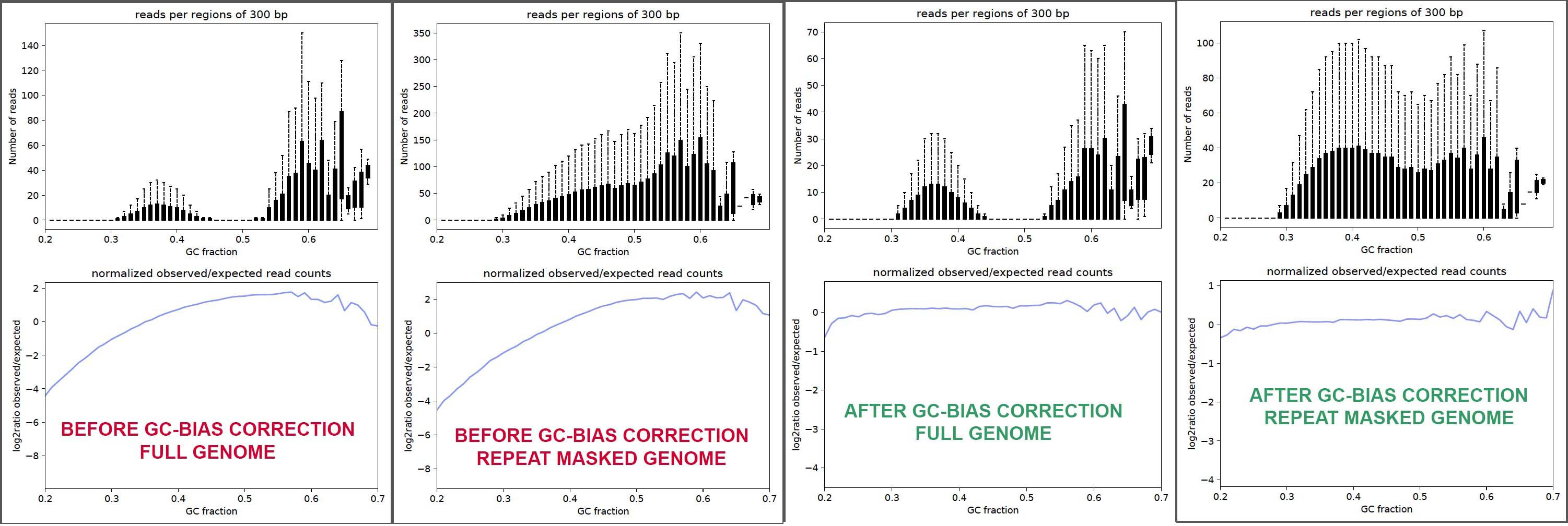
- There is GC-bias in my input sample (red font marked plots), as seen in the bottom sub-panels, whether it be using the full genome or the repeat-masked genome.
- GC-bias correction works as expected in terms of smoothing log2(observed/expected) Vs GC% in the corrected output (green font plots)
- Post-GCbias correction, it looks like for full genome case, reads in the >60% GC-range have been down-sampled.
- I suppose it does not make sense to examine reads against the masked version of the genome because of the possibility that expression can truly be from repeat regions, and other reason(s) ?
- Should I use GC-bias corrected BAM file with featureCounts to report gene expression as count data?
- OR Is this step ONLY to examine and acknowledge if there is GC-bias, but use other preferred methods to compensate for GC-bias?
- If latter, then which bioinformatic protocol(s) to follow for factoring in GC-bias at a later stage in my analyses?
gc-bias
deeptools



