Hello,
I am very new to the world of sequencing and would really appreciate your knowledge. I am trying to study a genomic region containing 5 very homologous genes and have obtained the FastQ files generated using MinION. I used the tool NanoPlot to produce a QC report but am struggling in understanding it. Particularly regarding the quality scores and quality cut-offs; I appreciate a quality threshold depends upon my application but would it be possible to explain very simply what the number of the 'mean read quality' actually represents? If I go on to filter my reads, is there a standard cut-off for ONT data?
Any information around this would be extremely helpful!
 •
written
by
s.m.frampton •
•
written
by
s.m.frampton •

1 answer
TLDR: if you can align the reads (i.e. if you have a reference genome) then you might want to filter on mapping quality, and not on read accuracy.
In NanoPlot, the mean read quality is the mean of the base call quality scores. To be entirely correct, those (Phred scale) base call quality scores are first converted to their probability of being correct, averaged, and then back to the Phred scale. A bit more about that can be found in this blog post.
ONT will by default filter on a minimal quality score of 7, but that's quite arbitrary. I don't know why they went with that score. For my applications, which is structural variant detection, I don't filter at all. If I get a low-quality read, which does map reliably, and identifies a variant then I'm happy. The quality scores match quite well with the percent identity if your data is recent at least. So as such, you can estimate the error rate of a read, based on the quality score. A quality score of 7 corresponds with a ~80% accurate reads, which is not amazing. See also the image below for how the Phred score corresponds to the probability of error or the accuracy:
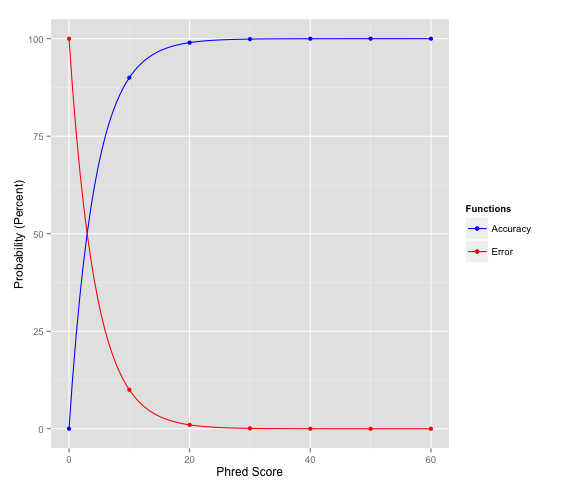
For your application I would let the aligner judge reads: if it aligns with a high mapping quality to one of those genes, and not to the others, then that's a good thing right? I don't know how similar the genes are, and how long your reads, though, and also not what your aim is.

Log in to answer this question.
-
ONT MinION QC: Concatenate or Analyze Individually
written by Acervo •
-
FastQC on nanopore data: high proportion of polyA and polyG. Why ?
written by Matt •
-
how to choose an efficient filtering threshold to remove shorter and low quality reads before doing…
written by Shaghayegh •
-
Using Nanoplot to asses quality of ONT data
written by matt81rd •
-
Interpreting EggNOG Results
written by Mason •
-
Degenerate Primer Sequence Recovery
written by james.nolan.2 •
-
Determining the PHRED Quality Score
written by adhamzul •
-
How to trim Nanopore reads. Please suggest a tool.
written by SUDOsundu •
-
Nanopore data workflow for detecting variance
written by s.m.frampton •
-
Extracting imputation (INFO) quality scores for specific SNPs from VCF files
written by e.jabbari •









Wikipedia is also not bad here, but it might be tricky to find the right page.
https://en.wikipedia.org/wiki/FASTQ_format
A rough estimated might by PHRED quality scores of ~8-12 for raw nanopore reads and ~30-35 for illumina reads.
Hello!
I have obtained a 'Median read quality' value of 9.4 for a metagenomic sample through Nanoplot analysis. Is there any mathematical formula for converting this value to a Phred score?
Regards
9.4 is the phred score
Thanks a lot, sir!
Here is a snapshot of another part of my NanoPlot result:
Number, percentage and megabases of reads above quality cutoffs
Please suggest:
Thanks and Regards