Am I correct in understanding that you want to avoid calling significant the tests with p-values from the mode among the larger p-values? If so then consider replacing the BH-FDR procedure with a step-down procedure
First, suppose psi_m(i, delta) is some criterion function which is increasing, takes the value 1/m at i=1 and the value 1 at i=m. BH-FDR uses the criterion function i/m . The extra parameter, delta, is missing from the BH-FDR procedure but its meaning will become clear below.
A step-down procedure, using criterion function psi_m, is defined by the following expression for the number of significant calls
[***] R = inf{ i : P_(i:m) > psi_m(i, delta) alpha } - 1
and the particular test statistics called significant are the indexes (i:m) corresponding to the ordinals
1 <= i <= R
Contrast this to the step-up procedure, which results the following number of significant calls
R' = max{ i : P_(i:m) <= psi_m(i, delta) alpha } (note that for psi_m(i,delta) = i/m this is the BH-FDR procedure)
NOTE: for a given criterion function, psi_m, the step-down procedure is always more conservative than the step-up procedure.
A nice step-down procedure which actually controls the FDX (probability that the FDP exceeds a given value) is one due to Lehman and Romano defined by applying formula [***] above, with
psi_m(i, delta) = (floor(i delta) + 1)/(m + floor(i delta) + 1 - i)
This guarantees that
P( V/R > delta ) < alpha
and will result, for sufficiently small alpha, in calling significant only test statistics corresponding to p-values from the mode to the left. A reasonable choice for delta would be delta = alpha
My R-package, pwrFDR, has relevant tools for applying the Romano procedure as well as tools for deriving sample size and power (determined via average power or via the TPX)
https://cran.r-project.org/package=pwrFDR
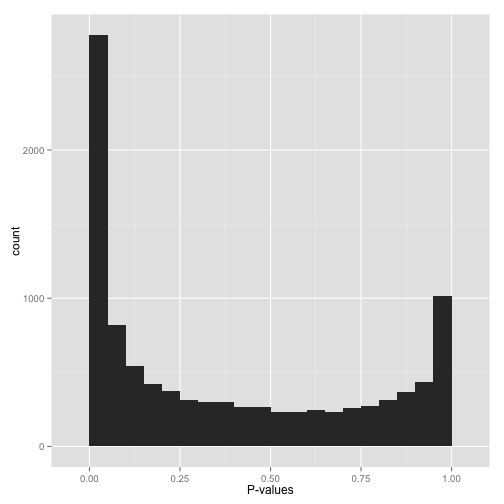












Hi, A nice explanation is given here http://varianceexplained.org/statistics/interpreting-pvalue-histogram/ . The right side could be due to lack of data. Having a cutoff before testing (like minimum number of reads could help) .
Hi microfuge,
That article is actually how I realized that this non-uniform distribution was an issue. I think that applying a correction when the values are non-uniform just makes the estimate more conservative from what I've read and simulated in R. Given that I'm comparatively okay with more Type-II errors compared to the same number of Type-I errors, I'll probably apply the methods as is since I don't want to do too much a posteriori database cleanup if possible.
Oh Sorry. I posted in a hurry and missed the last line specifying the image source. Makes sense.
No problem! That was actually one of the only websites that I could find that goes into any real detail on this issue. It kind of makes me concerned for everyone who has corrected a set of p-values for multiple hypothesis testing without checking the histogram.