Entering edit mode

5.5 years ago
nazaninhoseinkhan
▴
520
Hi all, I want to visualize my copy number alteration results following annotating with Variant Effect Predictor (VEP) as a CNV plot.
The input of VEP had been generated by RgenomicRanges package in R. I found several significant deletions or duplications (qvalue<0.2)
Here is what I obtained from VEP:
#Uploaded_variation Location Allele Consequence IMPACT SYMBOL Gene Feature_type Feature BIOTYPE EXON INTRON HGVSc HGVSp cDNA_position CDS_position Protein_position Amino_acids Codons Existing_variation DISTANCE STRAND FLAGS SYMBOL_SOURCE HGNC_ID TSL APPRIS SIFT PolyPhen AF AFR_AF AMR_AF EAS_AF EUR_AF SAS_AF AA_AF EA_AF gnomAD_AF gnomAD_AFR_AF gnomAD_AMR_AF gnomAD_ASJ_AF gnomAD_EAS_AF gnomAD_FIN_AF gnomAD_NFE_AF gnomAD_OTH_AF gnomAD_SAS_AF CLIN_SIG SOMATIC PHENO PUBMED MOTIF_NAME MOTIF_POS HIGH_INF_POS MOTIF_SCORE_CHANGE
11_66333072_deletion 11:66333071-66333838 deletion coding_sequence_variant,intron_variant,feature_truncation MODIFIER CTSF ENSG00000174080 Transcript ENST00000310325 protein_coding 5/8/2013 5/8/2012 - - 755-? 645-? 215-? - - - - -1 - HGNC 2531 - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
11_66333072_deletion 11:66333071-66333838 deletion coding_sequence_variant,intron_variant,feature_truncation MODIFIER CTSF ENSG00000174080 Transcript ENST00000524994 protein_coding 3/6/2009 3/6/2008 - - 187-? 189-? 63-? - - - - -1 cds_start_NF,cds_end_NF HGNC 2531 - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
13_50135463_deletion 13:50135462-50141345 deletion coding_sequence_variant,intron_variant,feature_truncation MODIFIER RCBTB1 ENSG00000136144 Transcript ENST00000258646 protein_coding 1/2/2011 1/2/2010 - - 115-? 71-? 24-? - - - - -1 - HGNC 18243 - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
I also tried to perform copy number alteration analysis on TCGA data using Gistic 2.0 in GenePattern. However stragely no amplification or deletion were detected.
Can you tell me why I did not get significant results when running gistic 2.0.
Regards
Nazanin




See: How to add images to a Biostars post
VEP and GISTIC are apple and orange, IMO. Not clear how the analysis is done. Did you try other CNA tools using CBS or Haar algorithms?
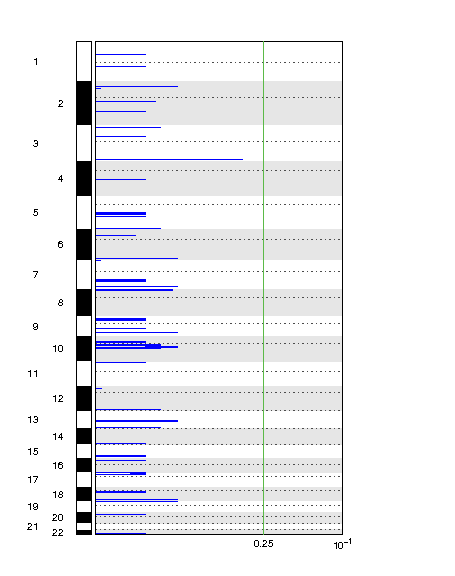
Do you have an example image for Deletion depiction?
This is the link: https://postimg.cc/yDv16Z8q
Nazanin, as I understood from the previous question, you only wanted to use VEP for the purpose of annotating the regions. VEP is usually the end-point in an analysis, i.e., the final step.
Going back to this step: C: Annotation of huge number of CNV files
Were you ever able to run that command (
runGAIA()) successfully? It will likely not work on any personal computer because it is too compute intensive.Again, the pipeline that works is this:
Yes Kevin, you're right.
If you remember after getting the result of copy number analysis I tried to run biomaRt as you suggested to me. However I could not run it properly so I decided to use VEP instead. I have access to server right now, however I am a little confused about how I can generate a custom CNV plot to represent in my paper.
I tried to re-perform the analysis using gistic 2.0 on Genepattern, however no significant results was generated. This made me more confused.
Can you possibly retry the annotation step with biomaRt? The exact steps are here: A: How to extract the list of genes from TCGA CNV data
The previous thread became too long...
Sure. I will rerun biomaRt on Monday and let you know at which step I got the error message. I think the source of problem is what I introduce to biomaRt as input.
Thank you again
I have also just posted an answer below, showing how you can plot output from runGAIA. Let me know if it works.
If you need me to process the data for you on my servers, then I will do that - no problem. Just point me to which Firebrowse data you are using. You can contact me in private, if you wish.
Sure Kevin.
I will inform you on Monday.
Hi cpad, the original thread had dragged on, here: C: Annotation of huge number of CNV files
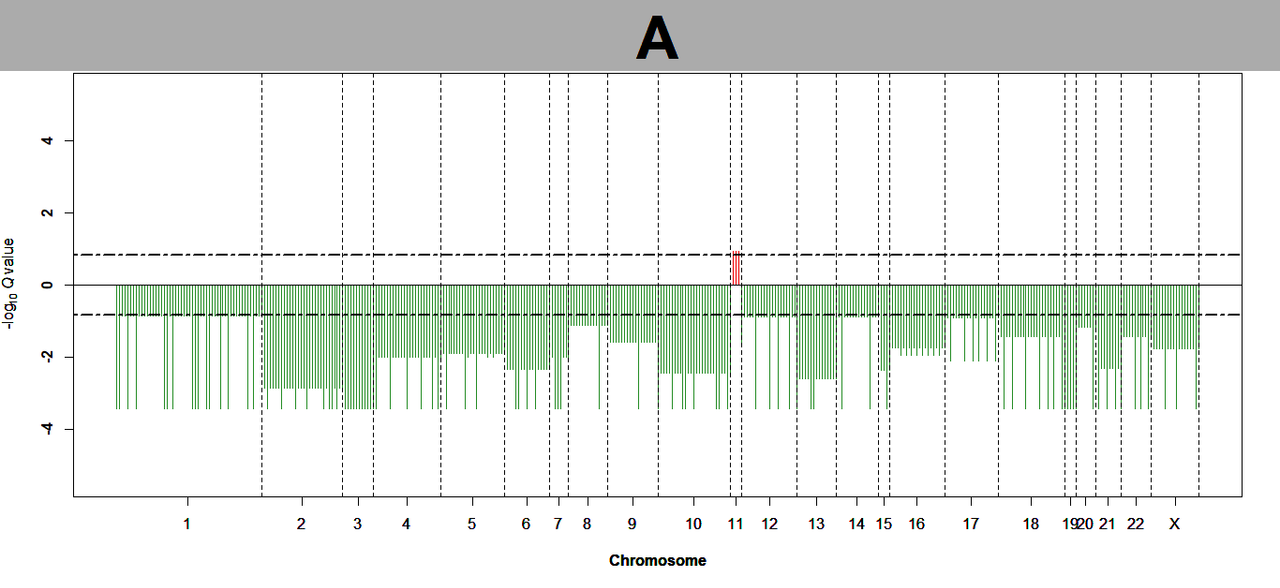
Hi @Kevin Blighe I am getting a similar plot like this one, all commands seem to run without any errors, exactly same steps followed form part I, part II and part III. Could you help with it?
Thank you
Do not add answers unless you're answering the top level question. "I have this problem too" comments should be comment-replies on appropriate posts, not answers. I've moved your post to a comment for now, but please be more careful in the future.