Entering edit mode

5.6 years ago
Gina
▴
10
Hi,
How can I find out how many protein-encoding genes I have in my sequence?
Hi,
How can I find out how many protein-encoding genes I have in my sequence?

You could try with the EMBOSS command showseq. For instance:
echo atgtttcaggacccacaggagtaa | showseq -filter -threeletter y -format 4
10 20
----:----|----:----|----
atgtttcaggacccacaggagtaa
MetPheGlnAspProGlnGlu***
But you can get the all three/six reading frames and single code:
echo atgtttcaggacccacaggagtaa | showseq -filter -format 5
10 20
----:----|----:----|----
atgtttcaggacccacaggagtaa
M F Q D P Q E *
C F R T H R S X
V S G P T G V X
echo atgtttcaggacccacaggagtaa | showseq -filter -format 6
10 20
----:----|----:----|----
atgtttcaggacccacaggagtaa
M F Q D P Q E *
C F R T H R S X
V S G P T G V X
----:----|----:----|----
X N * S G C S Y
X T E P G V P T
H K L V W L L L
On Linux platforms, EMBOSS is installed with sudo apt install emboss.
but isn't looking for ORF the procedure for start characterizing a genome? Isn't the standard approach something like this figure (taken from Danos et al. EMBO J 1982;1(1):231)?
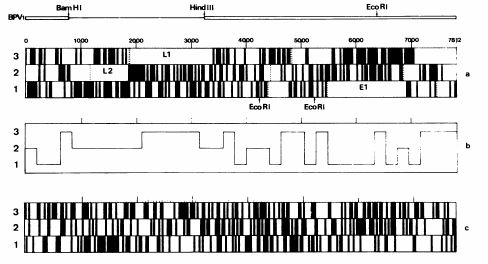
Does the procedure give an exact number? No, it is (both in the figure than with showseq) a graphical representation, but I think is a beginning.
I assumed the sequence was given as fasta format; should one have a GenBank format, it would be possible to get the number of genes with something like (on a unix terminal):
grep 'gene.*[0-9]*\.\.[0-9]' <file>
For instance, saving the data for NC_009333.1 (Human herpesvirus 8) on hhv8.gb, I get:
$ grep 'gene.*[0-9]*\.\.[0-9]' hhv8.gb | wc -l
96
For simple genomes (where one does not expect genes to be composed of exons/introns) a simplistic approach of looking for ORF is fine. Even then not every open reading frame would be expected to code for a protein. Which is the reason methods using HMM's were developed (even for prokaryotes) to predict coding regions.
Use of this site constitutes acceptance of our User Agreement and Privacy Policy.
This sounds like an assignment. If so you should state that clearly. Have you been asked to write a program or just do this using
blast(since you included that as a tag)?Problem description seems clear enough so you should be thinking of solving the questions on your own. Since you don't have access to your input data you have to go forward on your own.
Depends on what organism the sequence is from. For Eukaryotes you may need to use a gene prediction tool like GENSCAN, EGPRED etc. For prokaryotes you could simply look for start/stop sites and/or use a gene prediction tool like GLIMMER. The predictions can then be followed by sequence searches to identify the genes. If you don't need to precisely predict gene models then a simple blast search may suffice.
Edit: I am going to keep my original comment since you edited the original post.