Entering edit mode

7.0 years ago
Cutting DNA into small fragments is a key preparation step for DNA sequencing with NGS technology. To reduce errors and increase reliability of the sequence information, every genomic region should be sequenced several times. This means that several copies of a target DNA have to be cut in different ways to produce overlapping fragments to ensure an good coverage of the whole region of interest. This approach is based on the general idea that genomic DNA break-points are random and sequence-independent.
But there is a problem... read more
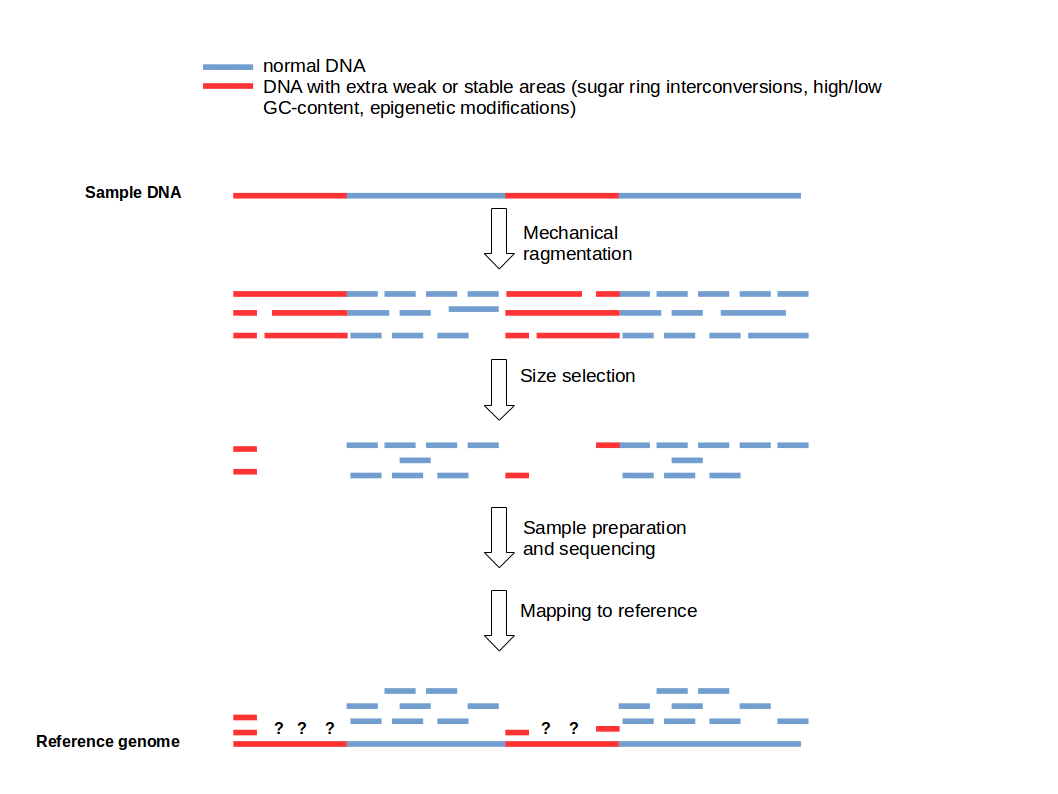
Funny, we were talking about this internally today. It's certainly the case that open vs. closed chromatin will react differently do sonication (or similar) and therefore produce different fragment size distributions due to the same treatment. Given the size selection that occurs during library prep. it "should" then be unsurprising to see regions enriched/depleted accordingly...but it seems that people always conveniently forget that.
Always good to see reminders like this.
About 3 years ago my lab had planned to do an experiment where DNA was sonicated in such a way that well-sonicated fragments (100-700bp) would be sequenced, and compared to poorly sonicated fragments (10kb+) that underwent an additional step of mild-decrosslinking and re-sonicated to get it to the 100-700bp range. Before I got the go-ahead to do the experiment though, someone else apparently published the answer, and found no difference between the two populations. In essence, while it doesn't make a whole lot of sense to me why these two populations of chromatin would be the same, data is apparently out there that shows that they are :-/
An argument could be made that the type of crosslinking performed (paraformaldehyde, formaldehyde, with/without methanol, only methanol, no fixative at all, etc) is more of a factor than sonication intensity/frequency/duration, at least from the experiments i have done. At Epigenetic conferences, chemists would routinely comment on why the field uses formadehyde derivatives rather than more modern fixatives, but no one was ever particularly interested in doing a study of enrichment for different fixatives, particularly as these questions were frequently, unfortunately, received as "have you considered using [my favourite fixative]", in much the same way questions like "have you looking at [my favourite gene]" often are. I'd be surprised if there isn't something published on this though.
People are much more focused on single-cell now anyway, so it could be another 3 years until this question comes up again :P
I wouldn't be surprised if more/less aggressive crosslinking will change the answer. This came up internally due to someone noticing that input samples from Encode and DEEP (made using NEXSON) have different profiles around the TSS. Encode input samples show enrichment around the TSS, those made with NEXSON show a mild depletion of signal around the TSS. The thought was that this is due to differences in sonication and fragment size distributions (NEXSON produce 100-800 base fragments while Encode used much much longer fragments). A difference of response to sonication would then lead to the profiles we and others observed.
Hm, very interesting...
Perhaps take a look at the adipocyte DEEP samples, as these samples underwent almost all the steps of NEXSON (in that Laura sequenced them like all the other DEEP samples, and i sonicated the purified nuclei with something very close to Laura's sonication settings), but purification of nuclei was not done using sonication (the big benefit of NEXSON) but by old-school douncing. If the adipocyte samples are closer to ENCODE than the other DEEP samples, then it would suggest the part of NEXSON that is responsible for this phenomena would be the lysis-by-sonication step, and you can exclude all the factors that were the same between the adipocyte samples and, say, the DEEP hepatocyte samples which were done around the same time with all the same processes. You guys are probably already on top of it though
Crosspost on SEQanswers