
This might sound a very basic question to some of you, but for me it has turned to a dilemma.
I am trying to align MiSeq deep sequencing targeted amplicons to human genome (paired ended).
This is the way I do this:
- I check the quality and trim for bad quality and adapter sequences
- I align the reads using bwa aln algorithm
- Convert the SAI files to SAM (using bwa sampe)
- Convert SAM to BAM (using samtools)
- Sorting and indexing the bam files (using samtools)
- Run GATK's indel realigner
- Update BAM files header
I used to do this all the way up until step 5 and recently I added steps 6 and 7 but the results still look the same.
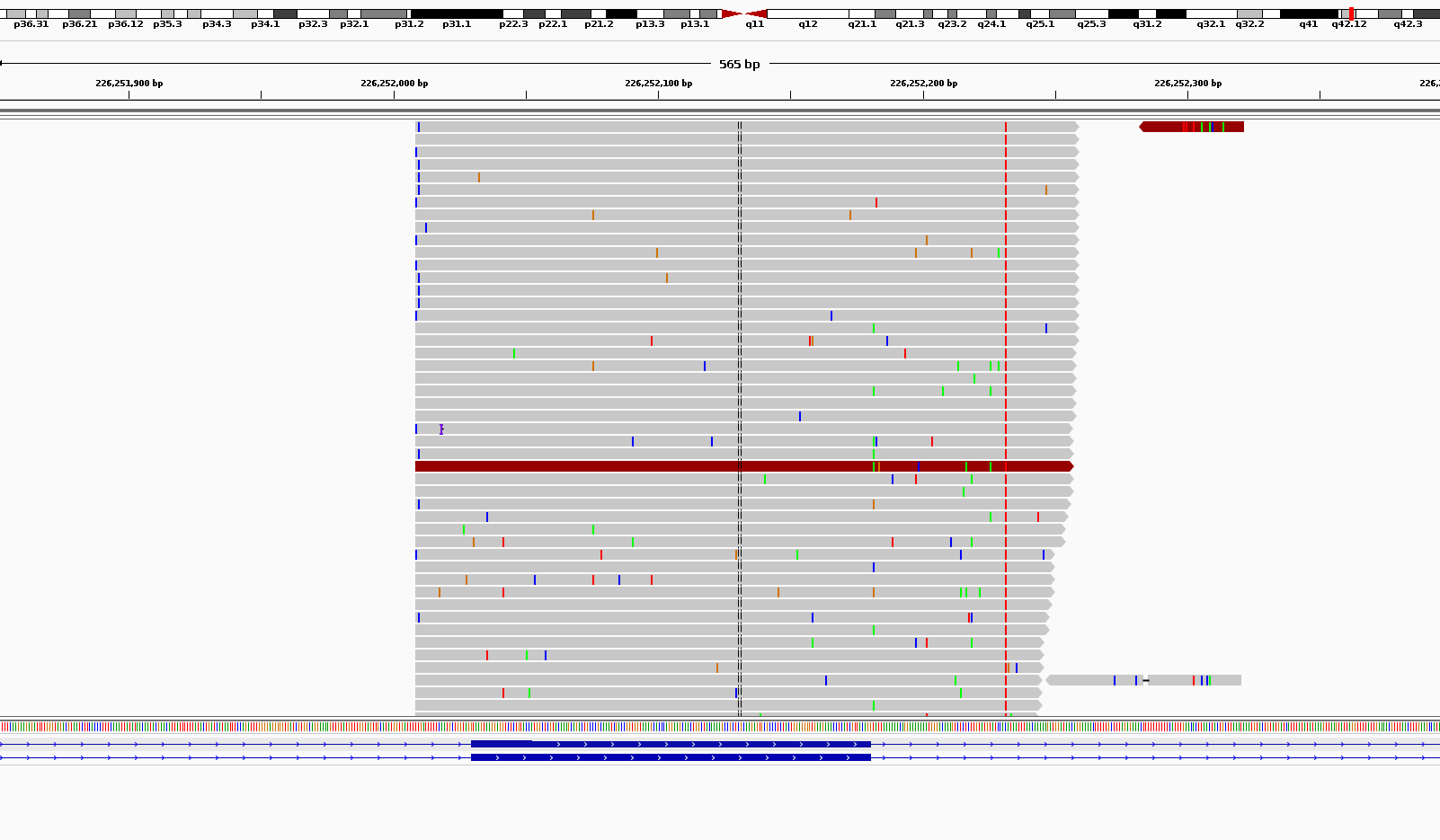
What I get is somehow weird in terms of noises. As you can see in the attached picture for coverages around 1000x I get scattered noises all around my reads.
Am I doing something wrong or this is the what it is supposed to loo when we do ultra-deep sequencing?
Any thoughts will be appreciated.
UPDATE:
Apparently doing steps 6 and 7 doesn't do any good and it actually messes up some of the aligned reads! This I came across when I compared some samples gone through 1-5 and 1-7 steps.



Hi Nikleotide can you share your code? Thanks